

Студопедия — теоретические основы. окисление аммиака до окиси азота является начальной стадией производства азотной кислоты.
Окисление аммиака до окиси азота является начальной стадией производства азотной кислоты.
Наилучшим избирательным действием на процесс окисления аммиака до окиси азота обладает платина, которая при высоких температурах может давать выход до 99 %.
При проведении процесса на платиновых катализаторах окисление аммиака начинается уже при 145°С с получением главным образом элементарного азота и закиси азота. Наибольший выход окиси азота 95-98 % достигается при температурах 850-950 ° С.
При повышении температуры возрастают потери платины, что приводит к необходимости ограничить несколько верхний температурный предел процесса. Повышение температуры выше 1 000°С приводит к побочным процессам. Чрезмерное повышение температуры исходного газа сопровождается диссоциацией аммиака и окиси азота.
Оптимальные температуры окисления аммиака на платине при атмосферном давлении находятся в интервале 800-840°С; для окисленных катализаторов оптимальные температуры несколько ниже и составляют 700-800 °С.
В качестве окисляющего агента для процесса окисления аммиака используется в подавляющем большинстве кислород воздуха. Расход кислорода на окисление аммиака до окиси азота может быть определен согласно уравнению:
4NH3 5O2 → 4NO 6H2О
Таким образом, стехиометрическое мольное соотношение кислорода к аммиаку O2/NH3 составляет 1,25. Однако при таком соотношении выход окиси азота незначителен. Для его повышения необходим определенный избыток кислорода, от которого будет зависеть и концентрация аммиака в воздушно-аммиачной смеси.
В производственных условиях содержание аммиака в воздушно-аммиачной смеси поддерживается в интервале 9,5-11,5 %, что соответствует отношению O2/NH3 — 1,7-2,0. Для окисных катализаторов, имеющих меньшую, чем платина активность, соотношение O2/NH3 должно быть более двух и соответственно концентрация аммиака в воздушно-аммиачной смеси должна поддерживаться в пределах 7,5-9,5 %.
При изменении концентрации аммиака в исходной смеси следует учитывать, что воздушно-аммиачные смеси могут быть в определенном интервале взрывоопасны. Применяемые в промышленности аммиачно-воздушные смеси (9,5-11,8 %) практически не представляют опасности. Что касается аммиачно-кислородных смесей, то они воспламеняются со взрывом при любой концентрации аммиака в интервале температур 700-800 ° С.
Реакция окисления аммиака до окиси азота протекает во внешне диффузионной области как на платине, так и на окисных катализаторах. Сама реакция протекает быстро, но общая скорость процесса вследствие внешнедиффузионного торможения может снижаться более чем на 90 %.
Максимальной степени превращения аммиака в окись азота соответствует определенное время контакта.
Практическое осуществление процесса окисления аммиака требует таких условий, которые обеспечивают максимальное протекание основной реакции и минимальной потери аммиака в виде молекулярного азота. С этой целью окисление аммиака проводят в присутствии активного, избирательно действующего катализатора, в сравнительно узком интервале температур, при строго определенных времени соприкосновения газа с катализатором и начальном составе аммиачно-воздушной смеси.
Наиболее распространенным промышленным катализатором процесса является платина или ее сплавы с родием и палладием. Для обеспечения большой поверхности катализатора платиновые катализаторы обычно используются в виде сеток из проволоки диаметром 0,045-0,09 мм, причем свободное сечение сетки составляет 50-60 %. Достаточно полный контакт газа с катализатором достигается установкой в аппарате последовательно трех сеток.
Из всех металлов платиновой группы наиболее оптимальными являются добавки родия. Сплавы платины с родием характеризуются не только более высокой каталитической активностью, чем чистая платина, но имеют лучшие прочностные показатели. Особенно это важно при работе в области высоких температур, так как у них точка плавления сплава выше, чем у платины.
Применяются в промышленности также палладиевые катализаторы, несколько уступающие платинородиевым по механической прочности, но имеющие высокую каталитическую активность.
Различные сплавы платины и палладия с добавками родия, серебра, иридия, кобальта, вольфрама и др. обеспечивают выход окиси азота 96-99 %. Из неплатиновых катализаторов окисления аммиака наиболее активными оказались катализаторы на основе оксидов железа и кобальта; активированные добавки хрома, марганца, висмута, никеля и др.; многие из этих катализаторов показали высокую активность. Так, на окисножелезном катализаторе, промотированном окислами висмута и марганца, степень конверсии достигает 94 %.
Однако, для всех окисных катализаторов наблюдается потеря активности во времени. Для систем, работающих при атмосферном давлении, применяется двухступенчатое окисление аммиака: 1-ая ступень платинородиевая сетка, 2-ая ступень — слой таблетированного окисного (чаще железо-хромового) катализатора, толщиной 50-65 мм. Общий выход окиси азота в таких аппаратах 96,5 %, затраты на платину в этом случае сокращаются в 3 раза, снижаются и потери платины.
Все окисные катализаторы относятся к типу осажденных и готовятся совместным осаждением гидроокисей из растворов серно- и азотнокислых, хлористых солей. Неплатиновые катализаторы на носителях практически не применяются вследствие малой активности.
Согласно теории диффузионной кинетики окисления аммиака. Скорость процесса его окисления на платине определяется наиболее медленной стадией — диффузией аммиака к поверхности катализатора.
Продолжительность реакции окисления аммиака на платине составляет 10 -4-10 -5 сек. Время контактирования может быть определено из уравнения:
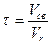
где Vсв — свободный объем катализатора, м3 /сек;
Vr — объемная скорость газа в условиях конверсии, м3 /сек.
Существует вполне определенное время контакта и соответственная ему скорость газового потока, обеспечивающие максимальный выход окиси азота.
Для осуществления реакции необходимо соответствующее энергетическое состояние реагирующей системы, определяемое значениями энергии активации.
Процесс каталитического окисления аммиака начинается со стадии активированной адсорбции кислорода на поверхности катализатора с образованием промежуточного соединения, затем происходит активированная адсорбция аммиака, требующая меньшей энергии активации; при этом образуется переходной комплекс с последующей перегруппировкой его в комплекс. Возможно образование других промежуточных соединений гидроксиламина NH2OH, нитроксила NHO, амида NH2, атомарного азота и др. и в конечном счете образование NO, N2, H2O.
Из всех термодинамически возможных реакций в присутствии катализатора в первую очередь протекает та реакция, которая требует наименьшей энергии активации. В соответствии с адсорбционно-химической теорией катализа механизм каталитического окисления на платине можно представить следующим образом. Кислород и аммиак диффундируют из потока к поверхности катализатора. Находящиеся на поверхности платины атомы со свободными валентностями обеспечивают первоочередную активированную адсорбцию кислорода за счет возникновения электронной связи. Химическое взаимодействие платины с кислородом приводит к ослаблению атомарных связей в молекулах и образованию перекисного комплекса катализатор — кислород. Последующая активированная адсорбция аммиака дает в результате комплекс катализатор-кислород-аммиак и в конечном результате происходит перераспределение электронных связей и соединение атомов азота и водорода с кислородом.
Адсорбированный кислород не входит в кристаллическую решетку платины и образует с ней непрочные связи. Молекулы аммиака ориентируются к кислороду атомами водорода (вследствие высокого сродства кислорода и водорода) с последующим образованием молекул окиси азота и воды. NO и Н2О свойственна малая адсорбционная способность; вследствие этого они десорбируются с поверхности катализатора, освобождая связи для вновь сорбируемых молекул кислорода.
Механизм окисления аммиака на окисных катализаторах, по-видимому, не будет иметь принципиального отличия от такого на платине; однако, оптимальные условия процесса (температура, состав газа, время контактирования) в этом случае изменяются в зависимости от стадии, лимитирующей процесс в целом.
Целью данной работы является испытание активности неплатиновых катализаторов на лабораторной модельной установке в зависимости от условий: температуры, объемной скорости газа, его начального состава, времени контактирования и т.д.
§
Контактный аппарат представляет собой кварцевую или фарфоровую трубку с катализатором 1, установленную в вертикальной (или горизонтальной) трубчатой печи 2 с электрообогревом. Температура в контактном аппарате заменяется термопарой — 3 и регулируется автоматическим терморегулятором 4. В кварцевую трубку помещён неплатиновый гранулированный катализатор. Трубка имеет длину около 300 мм и внутренний диаметр 20-30 мм. Газ подается в контактный аппарат сверху через стеклянную трубку 5.
Газовая смесь готовится следующим образом: аммиак из склянки 6 через реометр 7 подается для смешения в стеклянный или алюминиевый смеситель 8 с воздухом, подаваемый воздуходувкой 9 через реометр 10.
Из смесителя газ подается в контактный аппарат и проходит в нем сверху вниз. Для поглощения окислов азота газ после контактного аппарата пропускается через поглотительную склянку 11 с раствором перекиси водорода и отводится далее в тягу.
Опыт проводится в следующем порядке: расходы аммиака и воздуха, требуемые для получения смеси заданного состава, устанавливают по заранее градуированному реометру. Общая объемная скорость газовой смеси задается в пределах 200-1000 см3/мин. Состав газа устанавливают и контролируют путем анализа. Газовая смесь с определенной заданной скоростью подается в контактный аппарат, нагретый до заданной температуры. Температура задается в пределах 600-850 ° С. Во время опыта скорость газа и температура в контактном аппарате поддерживается строго постоянной. В течение опыта 2-3 раза, через каждые 10 минут, отбираются пробы газа для анализа содержания аммиака.
Таким же образом проводятся опыты при других заданных условиях. Измеряемыми параметрами могут быть состав исходного газа, температура, время контактирования и состав исходного катализатора.
Для каждого данного катализатора при измерении одного из факторов технологического режима остальные условия опыта должны быть строго постоянными. Время контактирования можно изменить либо путем изменения скорости газа, либо высоты слоя катализатора. Время контактирования рассчитывается исходя из объемной скорости газового потока и свободного объема контактной зоны VCB.
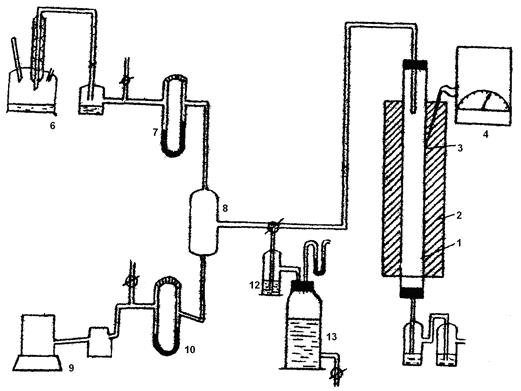
Рисунок 5 — Схема лабораторной установки окисления аммиака
1 – трубка с катализатором; 2 – трубчатая печь; 3 – термопара; 4 — терморегулятор; 5 – стеклянная трубка; 6, 11, 12 – склянки; 7, 10 – реометры; 8 – смеситель; 9 – воздуходувка; 13 – газометр
Исходная газовая смесь анализируется на содержание NH3 пропусканием газового потока при помощи аспиратора и крана через поглотительную склянку (дрексель), в которую предварительно заливают 50 см3 воды, 10 см3 0,1 н раствора НС1 и несколько капель метилового оранжевого. Поглотительная склянка соединена с аспиратором, создающим разряжение для протягивания газа через поглотительную склянку. Газ пропускается через поглотитель до тех пор, пока красная окраска раствора не перейдет в желтую.
Количество воздуха в отобранной на анализ пробе аммиачно-воздушной смеси определяют по объему воды, вытекающей из аспиратора. Содержание аммиака в газе (в объемных %) вычисляется по формуле:
 (5.1)
(5.1)
где а — объем NH3, см3,
V — объем вытекающей воды из аспиратора, см3
Р1 — давление по барометру, мм.рт.ст.,
t — температура в помещении, ° С,
Р2— упругость водяных паров при температуре, мм.рт.ст.
Анализ газа после контактирования проводится методом эвакуированных колб. В колбу заливают 5 см3 3 %-ого раствора перекиси водорода и, поместив колбу в матерчатый мешок (или обернув ее полотенцем), присоединяют с помощью резинового шланга к вакуумнасосу: колбу вакуумируют до остаточного давления 200-300 мм рт. ст., проверяют герметичность шлифа колбы; для этого колбу переворачивают горлом вниз и наблюдают, не происходит ли всасывания воздуха (если колба не герметична, то видны пузырьки воздуха, проходящие через раствор перекиси водорода). Если шлиф колбы не герметичен, то следует вынуть пробку со шлифом, протереть горло колбы насухо (фильтровальной бумагой), смазать его вакуумной смазкой, протереть пробку со шлифом в горловине и вновь вакуумировать. Далее колбу взвешивают на аналитических весах и засасывают в нее анализируемую газовую смесь, присоединяют колбу к газоотборнику. Перед взятием пробы сливают конденсат и продувают анализируемую газом трубку газоотборника.
Отбор газа в колбу проводят осторожно, проворачивая трубку со шлифом до совмещения отверстий в шлифе с отверстием газоотборного отростка горловины колбы. При повороте колбы следят за уровнем жидкости в манометре, на реометре — воздуха, прикрывая и открывая вновь отверстия для газоотбора, с учетом того, чтобы не происходил переброс жидкости в реометр. Отбор газа производят до прекращения колебаний жидкости в манометре, т.е. выравнивая давление в колбе и установке. После взятия пробы газа, колбу взвешивают снова и определяют навеску газа по разности.
Затем колбу встряхивают в течение 15-20 минут. При этом кислород воздуха, имеющийся в колбе, и перекись водорода окисляют окислы азота в азотную кислоту, которую далее оттитровывают раствором едкого натра. С этой целью содержимое вакуумированной колбы переливают в коническую колбу, куда добавляют и промывные воды от промывания вакуумированной колбы несколькими порциями дистиллированной воды.
Перед титрованием необходимо разрушить избыток перекиси водорода непродолжительным кипячением; перед кипячением к анализируемому раствору добавляют отмеренное количество титрованного 0,1 н раствора едкого натра. Количество едкого натра берут по заданию преподавателя в избытке в зависимости от концентрации газа и его навески. Избыток едкого натра оттитровывают после кипячения и охлаждения пробы 0,1 н раствором НСl с метиловым оранжевым — в качестве индикатора.
Количество едкого натра, израсходованное на нейтрализацию азотной кислоты, пересчитывают на окисленный аммиак.
Расчет результатов анализа производится по формуле:
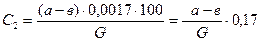 (5.2)
(5.2)
где C2 — содержание окисленного аммиака в конечном газе, % вес.;
а — количество см3 0,1 н раствора NaOH, прилитое к анализируемому раствору;
в — количество см3 ОД н раствора НС1, израсходованного на титрование избытка NaOH;
(а-в)0,0017 — количество аммиака, окисленного до NO в пробе газа, г;
G — вес пробы.
Для вычисления степени окисления X пересчитывают содержание аммиак в исходном газе также на весовые проценты по формуле:
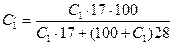 (5.3)
(5.3)
где С1 — концентрация NH3, % об.;
17 и 28 — соответственно молекулярные веса аммиака и воздуха.
Контрольные вопросы
1. Теоретические основы процесса окисления аммиака. Применение продуктов окисления.
2. Механизм окисления аммиака на окисных катализаторах.
3. Как определяется содержание аммиака в газе?
4. Анализ получаемого газа.
Литература
1. Исагулянц В.И., Егорова Г.М. Химия нефти. Руководство к лабораторным занятиям. 2-е изд. М., Химия, 1965, 517 с.
2. Лабораторный практикум по технологии основного органического синтеза/ Рейхсвельд В.О., Рубан В.Л., Саратов И.Е. М., Химия, 1966, 320 с.
§
Процессы этерификации, гидролиза, гидратации и дегидратации в промышленности основного органического и нефтехимического синтеза находят широкое применение для производства одноатомных и многоатомных спиртов, простых и сложных эфиров, одноосновных и многоосновных карбоновых кислот, ангидридов кислот, ненасыщенных соединений, фенолов, σ-оксидов и многих других ценных веществ. Кроме того, эти процессы часто являются промежуточными этапами в многостадийных синтезах других важных соединений.
Сложные эфиры применяют в качестве растворителей, пластификаторов, в производстве синтетических смазочных масел, гидравлических жидкостей, поверхностно-активных веществ, в парфюмерии и т. д. Сложные эфиры образуются в результате взаимодействия спиртов с карбоновыми кислотами, ангидридами и хлорангидридами карбоновых кислот:
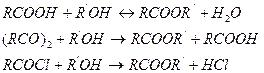
Если в этерификации участвует двухосновная кислота, то в зависимости от мольного соотношения спирта и кислоты может образоваться продукт как неполной, так и полной этерификации:
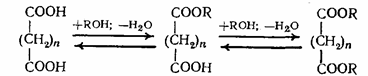
Полные и неполные эфиры образуются и тогда, когда взаимодействуют многоатомные спирты и одноосновные кислоты. Выход полных и неполных эфиров и в этом случае зависит от исходного мольного соотношения реагентов:
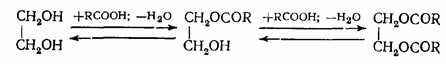
Если же кислоты и спирты содержат две и более функциональных групп, процесс протекает с образованием полиэфиров линейной или пространственной структуры.
Подавляющее большинство реакций этерификации являются равновесными, причем обратная реакция представляет собой гидролиз сложных эфиров. К классу этерификации относят и реакции алкоголиза, ацидолиза, переэтерификации, которые также равновесны:
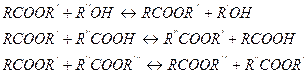
Все перечисленные реакции хорошо катализируются сильными кислотами (например, H2SO4, HC1) и только в случае алкоголиза сложных эфиров более эффективными катализаторами являются3 вещества основного характера (алкоголяты, щелочи и др.).
Спирты с карбоновыми кислотами взаимодействуют практически без выделения или поглощения тепла, и поэтому константа равновесия этих реакций мало зависит от температуры. На константу равновесия влияет как строение спирта, так и строение кислоты.
Существует несколько способов для смещения равновесия в сторону образования эфира. В жидкофазных процессах этерификации высокой степени конверсии исходных реагентов достигают при отгонке из реакционной массы воды или эфира, а если это невозможно, то повышают концентрацию одного из исходных компонентов. Последний способ применяют и в процессах газофазной этерификации.
Механизм реакции этерификации в присутствии протонодонорных кислот можно представить следующей схемой:
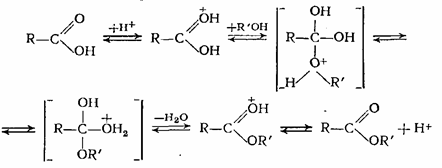
В этой системе равновесных превращений самой медленной является реакция спирта с протонированной кислотой, а в обратном направлении (при гидролизе или алкоголизе) реакция воды с протонированным эфиром.
Этерификация н-бутилового (пентилового) спирта уксусной кислотой
§
Уксусная кислота ледяная 30 г
н-Бутиловый спирт 45 г моль
Серная кислота концентрированная 0,5 мл (или катионит КУ-2 5 г)
Бензол (сухой) 150 мл
Карбонат натрия, 5%-ный водный раствор 100 мл
Сульфат натрия безводный 10 г
Бутилацетат (пентилацетат) получают на установке, схема которой приведена на рисунке 6. Установка состоит из автотрансформатора 1, электронагревателя 2, реактора 3, колонки 4, кожуха 5, головки 7, термометра 8, обратного холодильника 9 и сепаратора 10. Перед началом опыта проверяют правильность сборки и герметичность всех соединений установки.
Реактор изготовлен из термостойкого стекла и представляет собой круглодонную одногорлую колбу емкостью 300 мл. Из термостойкого стекла изготовлены также колонка, головка, обратный холодильник и кожух. Головка снабжена сепаратором 10 для отделения бензола от воды. Колонка заполнена стеклянной насадкой 6 на высоту 150 мм.
Наливают в реактор 30 г ледяной уксусной кислоты, 45 г н-бутилового спирта и капиллярной пипеткой на 1 мл добавляют0,5 мл концентрированной серной кислоты (или насыпают катионит КУ-2, подготовленный по методике — см. стр. 166). Для спокойного кипения реакционной массы в реактор опускают несколько небольших кусочков керамики. Реактор присоединяют к нижней части колонки с небольшим зазором в шлифовом соединении. Устанавливают в верхнюю часть обратного холодильника воронку и наливают в установку 150 мл бензола. Часть бензола остается в сепараторе головки, а основная его масса стекает через дефлегмирующую колонку в реактор.
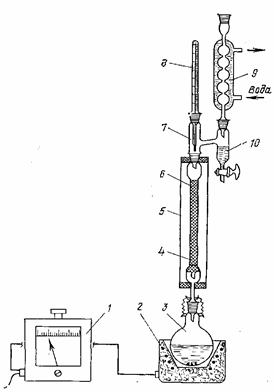
Рисунок 6 — Установка для этерификации с азеотропной отгонкой воды;
1 — автотрансформатор; 2 — электронагреватель; 3 — реактор;
4 — колонка; 5 — кожух; 6 — стеклянная насадка; 7 — головка; 8 — термометр; 9 — обратный холодильник; 10 — сепаратор.
Воронку удаляют, реактор плотно присоединяют к дефлегмирующей колонке и закрепляют шлифовое соединение металлическими пружинными скрепками. Подают воду в обратный холодильник и включают обогрев реактора. Температуру нагрева регулируют автотрансформатором таким образом, чтобы реакционная масса кипела умеренно, а скорость истечения конденсата из обратного холодильника в сепаратор составляла 1—2 капли в секунду.
Пары азеотропной смеси проходят через дефлегмирующую колонку, конденсируются в обратном холодильнике и стекают в сепаратор, в котором вода отделяется от бензола. Бензол снова перетекает в реактор, а вода собирается в нижней части сепаратора.
Конец реакции определяют по прекращению выделения воды из конденсата, стекающего из обратного холодильника в сепаратор, и по объему воды, скопившейся в сепараторе: он должен быть не менее 9 мл.
По окончании опыта выключают обогрев реактора, реакционную массу охлаждают до комнатной температуры, переливают в делительную воронку емкостью 300 мл и промывают последовательно 100 мл 5%-ного водного раствора карбоната натрия и 100 мл воды. После каждой промывки реакционной массе дают хорошо отстояться. Органический слой отделяют от воды, сливают в сухую коническую колбу, сушат над безводным сульфатом натрия, фильтруют в предварительно взвешенный на технических весах куб ректификационной колонки, определяют массу и перегоняют. В процессе ректификации ведут запись перегонки в таблицу и выделяют головную фракцию (бензол с остатками воды), промежуточную фракцию и фракцию бутилацетата. Определяют массы всех выделенных фракций и кубового остатка. Для фракции бутилацетата дополнительно определяют показатель преломления и эфирное число. Составляют материальный баланс опыта.
Контрольные вопросы
1. Сущность процесса этерификации.
2. Применяемые при данном процессе катализаторы.
Литература
4. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 171-176.
5. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с.
6. Паушкин Я.М., Вишнякова Т.П., белов П.С. Практикум по нефтехимическому синтезу. М.. Химия, 1965. 208 с.
7. Лабораторные работы по химии и технологии полимерных материалов. Пер. с польск. / Кухарский М., Линдеман Я., Мальчевский Я. И др. М., Химия, 1965, 396 с.
§
В настоящее время каталитический крекинг является важнейшим процессом нефтепереработки. Превращение нефтепродуктов в присутствии катализаторов позволяет получить с высоким выходом бензин с октановым числом до 85, керосино-газойлевые фракции (топлив для дизелей и газовых турбин) и газ, содержащий большое количество предельных и непредельных углеводородов Сз-С4, успешно используемый в промышленности органического синтеза. Избирательность катализаторов, зависящая от химического состава и структуры, определяет соотношение выходов различных продуктов (газ, бензин, кокс) и их состав.
В отличие от бензина термического крекинга, бензин каталитического крекинга характеризуется гораздо большей стабильностью вследствие отсутствия диеновых соединений и серы и обладает более высокой антидетонационной стойкостью, что объясняется также высоким содержанием изоалканов и ароматических углеводородов.
В данной работе следует определить выход основных продуктов в процессе каталитического крекинга индивидуальных углеводородов (изооктана) и провести их анализ.
Каталитический крекинг проводится обычно в паровой фазе при 450 -520° С давлении 0,1 — 0,2 МПа и продолжительности контакта в несколько секунд. В этих условиях каталитические реакции проходят гораздо быстрее реакций термического крекинга и являются более сложными. Наиболее важными, кроме реакций разложения, являются реакции изомеризации и перераспределения водорода, которые определяют высокое качество крекинг-продуктов, а также процессы уплотнения, ведущие к образованию кокса и его отложению на катализаторе. Скорость перечисленных реакций для отдельных классов углеводородов различна. Наибольшей реакционной способностью обладают олефино-вые углеводороды. Хотя каталитический крекинг относится к очень сложным процессам, все же начальные стадии процесса, т.е. образование первичных продуктов реакции (скорость распада индивидуальных углеводородов), могут быть приближенно вписаны уравнениями для режима вытеснения:
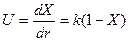 (7.1)
(7.1)
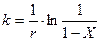 (7.2)
(7.2)
и уравнениями для режима смешения:
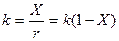 (7.3)
(7.3)
 (7.4)
(7.4)
Общая же константа скорости реакции будет определяться не только относительной скоростью распада предельных углеводородов, но и скоростью уплотнения непредельных углеводородов. При условиях, более благоприятных последней реакции, скорость крекинга будет подчиняться уравнении для реакции, протекающей между первым и вторым порядками.
Реакция крекинга осуществляется по радикально-цепному механизму, причем каталитический крекинг протекает через карбоний-ион. Самыми реак-ционноспособными карбоний-ионами являются третичные, способствующие образованию изопарафинов. Продукты каталитического крекинга отличаются своим составом и свойствами. Так в бензине имеется много изопарафинов и ароматических углеводородов, газ содержит большое количество изобутана и непредельных углеводородов, а газойлевые фракции отличаются высоким содержанием полициклических и ароматических углеводородов. В зависимости от фракционного состава сырья, состава и размера зерен катализатора, интервала применяемых температур процесс крекинга протекает в кинетической или переходной области. При диффузионном торможении крекинга в уравнение (1) вводится член, характеризующий замедление процесса, и константа скорости выражается уравнением:
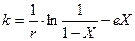
где е — постоянная, характеризующая степень торможения.
На выход и качество продуктов крекинга влияют следующие факторы: 1) вид сырья. 2) состав и активность катализатора. 3) температура и давление процесса, а также 4) объемная скорость подачи сырья в реактор и 5) продолжительность непрерывного крекинга без регенерации катализатора. При осуществлении каталитического крекинга применяют различные по составу и способу приготовления катализаторы. От качества применяемого катализатора, также как и от технологического режима процесса крекинга, зависит направление химического превращения топлива. Например, при применении катализатора, состоящего в основном из А1203 и Si02, происходит расщепление и полимеризация нафтеновых углеводородов. Мелкопористые катализаторы дают большие выходы газа, так как их поверхность менее доступна для молекул исходного сырья. К тому же крупнопористые катализаторы регенерируются легче, чем мелкопористые, при минимальной потере активности. Поэтому в промышленности предпочитают применять крупнопористые катализаторы, особенно для тяжелого сырья. Катализаторы для каталитического крекинга готовят из природных глин (типа флоридина) или синтетически. Так, аморфные алюмосили-катные катализаторы получают при совместном осаждении гидроокиси алюминия и окиси кремния из растворов соответствующих солей с последующей гидратацией. В последние годы широко применяются кристаллические алюмо-силикатные цеолиты с прочным трехмерным скелетом. В некоторых случаях они проявляют высокую активность без каких-либо добавок, но часто на них наносят активные компоненты.
Химизм превращения парафиновых углеводородов в ходе крекинга можно представить в виде основных реакций распада тяжелых парафинов и вторичных реакций изомеризации, циклизации, алкилирования и т. п. Так, для изооктана основные реакции распада:
С8Н18 →С4Н8 С4Н8
С8Н18 →С5Н10 С3Н8
С8Н18 →С6Н12 С2Н6 и т.д.
Продукты распада нафтенов также подвергаются вторичным реакциям изомеризации и дегидрогенизации с образованием ароматических углеводородов. В результате при каталитическом крекинге происходят передвижение кратной связи, изменение строения углеродного скелета, насыщение двойных связей, циклизация и алкилирование. Наибольшее значение в процессе крекинга имеет температура, определяющая степень и скорость каталитического разложения углеводородов, тем более что крекирующее действие катализаторов проявляется в сравнительно узком интервале температур. Повышение ее углубляет процесс разложения углеводородов. Так как процесс крекинга протекает в адсорбционном слое на поверхности катализатора, а не в объеме, то влияние давления незначительно. Повышение давления способствует полимеризации, перераспределению водорода и коксообразованию. Но в промышленности давление изменяют незначительно. На результаты крекинга влияет его продолжительность. Объемная скорость подачи жидкого сырья при каталитическом крекинге, как правило, изменяется в пределах от 0,1 до 10 дм3/ч ∙м3 катализатора. Наиболее употребительны объемные скорости 0,5-2,0 дм3/ч м3 катализатора при (0° С); чем меньше объемная скорость, тем больше глубина крекинга при прочих равных условиях.
Вследствие отложения кокса активность катализатора со временем падает и его необходимо регенерировать. Регенерация катализатора производится продуванием через него воздуха при 550-600° С, при этом кокс сгорает. Продолжительность непрерывного крекинга между регенерациями катализатора существенно отражается на результатах процесса. Она составляет от 1,5 до 10 мин. Чем меньше продолжительность работы катализатора, тем выше за этот период времени его средняя активность.
Источником образования кокса на катализаторе являются в первую очередь смолисто-асфальтовые вещества, содержащиеся в сырье. Регенерация катализатора, как правило, сложнее, чем проведение самого процесса крекинга. В ходе регенерации выделяется большое количество тепла от 25000 до 31000 кДж на 1 кг кокса. В области измеренных температур 450-500°С, регенерация протекает в кинетической области и ее скорость определяется скоростью реакции окисления. Но при повышении температуры до 500-550°С процесс регенерации переходит в диффузионную область.
§
Смолисто-асфальтовые вещества по сернокислотному способу определяют в отстойнике (рис. 1) — стеклянном цилиндрическом сосуде с притертой пробкой, суженном в нижней части. На этой части отстойника нанесена шкала с делениями по 0,5 мл.
Для анализа берут 10 мл «чистой» или «чистой для анализа» серной кислоты крепостью 95—96,5%, удельного веса 1,840. Серную кислоту следует держать в склянке с притертой пробкой; набирают ее оттуда пипеткой на 10 мл с надетой на нее каучуковой трубкой с зажимом. Отстойник должен стоять в деревянном штативе.

Рисунок 8. Отстойник для определения «акцизных» смол.
Кислоту осторожно, стараясь не разбрызгивать по стенкам, спускают в градуированную часть отстойника и закрывают его притертой пробкой. Так как мениск кислоты очень трудно заметить, то по стенке отстойника приливают немного бензина, тогда мениск кислоты отчетливо выделится на границе бензинового слоя.
В градуированный цилиндр емкостью 200 мл с притертой пробкой (в крайнем случае, притертую пробку можно заменить корковой) наливают 50 ли бензина, затем 50 мл тщательно высушенного испытуемого продукта и доливают бензина до метки 150 мл, т. е. еще 50 мл.
Для анализа применяют бензин «калоша» или бензин прямой гонки, выкипающий до 130°, очищенный серной кислотой.
Если продукт не просушен, то его обезвоживают измельченным безводным хлористым кальцием в количестве 5 г. Смесь 3 мин. взбалтывают и дают ей отстояться. После этого осторожно сливают в другой цилиндр емкостью 200 мл так, чтобы туда не попали кусочки хлористого кальция. Первый цилиндр споласкивают четыре раза чистым бензином (по 10 мл каждый раз), сливают его во второй цилиндр, где находится смесь, и доливают во второй цилиндр чистый бензин до метки 150 мл.
Покончив с приготовлением бензинового раствора продукта, открывают пробку отстойника и осторожно по стенке приливают к кислоте смесь из цилиндра. Цилиндр ополаскивают 5—10 мл бензина и сливают его в отстойник. Отстойник закрывают пробкой и, вынув его из штатива, сильно встряхивают два раза. После каждого встряхивания нужно обязательно открывать пробку для выпуска газов, образующихся вследствие испарения легких частей бензина, так как при действии серной кислоты смесь разогревается.
Отстойник со смесью энергично перемешивают 3 мин.; во время перемешивания его необходимо держать в горизонтальном положении и медленно вращать. После окончания взбалтывания устанавливают отстойник строго вертикально в штатив для отстаивания образовавшихся смолистых веществ от бензинового раствора продукта. По истечении 1 часа отсчитывают количество миллилитров черного смолистого вещества (кислый гудрон), прибавившегося к прежнему уровню кислоты. Это число, умноженное на 2, соответствует процентному содержанию смолисто-асфальтовых веществ, найденных по сернокислотному способу («акцизных» смол).
Если слои плохо разделяются, то в прибор осторожно по стенкам приливают вазелиновое, медицинское, трансформаторное или турбинное масло, которое образует слой между кислым гудроном и бензиновым слоем. Если стенки сосуда настолько забрызганы кислым гудроном, что не будет видно масла, то стеклянной палочкой осторожно счищают со стенок нижней части отстойника приставший к ним гудрон и после того, как граница обнаружится, яснее проводят отсчет.
При содержании в продукте от 20 до 50% асфальто-смолистых веществ его разбавляют в отношении 1:1 осветительным керосином, наливают в цилиндр 50 мл этой смеси и ведут работу по способу, описанному выше. Для нахождения процентного содержания асфальто-смолистых веществ полученное при отсчете количество миллилитров кислого гудрона надо умножить на 4.
При содержании в продукте более 50% асфальто-смолистых веществ испытуемый продукт разбавляют керосином в отношении 1:2. Для нахождения процентного содержания асфальто-смолистых веществ полученное при отсчете количество миллилитров кислого гудрона в этом случае надо умножить на 6.
1. Обработка результатов
Согласно ГОСТ 2550-44 количество асфальто-смолистых веществ на безводный нефтепродукт пересчитывают по формуле:
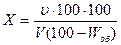
где v — приращение объема нижнего слоя в мл;
V — объем нефтепродукта, взятого для определения, в мл,
Wоб— содержание воды в нефтепродукте в % объемн.;
X—количество акцизных смол в безводном нефтепродукте в %.
Расхождение между двумя параллельными определениями не должно превышать одного деления градуировки отстойника. При достаточно внимательной работе расхождение между параллельными определениями обычно не превышает 0,4—0,5%.
При определении «акцизных» смол в продуктах, трудно подвижных при комнатной температуре, их необходимо подогреть.
Контрольные вопросы
1. Недостатки процесса сернокислотного способа определения смолисто-асфальтеновых веществ.
2. Почему в данном случае применяется серная кислота?
Литература
1. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с.
§
Реакционным сосудом для проведения гидратации ацетилена служит «утка» -1, закрепленная в установке для встряхивания (рисунок 10.2). Перед проведением опыта холодильники — 2 для поглощения ацетальдегида заполняются дистиллированной водой на 2-3 шарика и к кожуху холодильников подключается вода для внешнего охлаждения. Для проведения опыта в «утку» вносится 15 мл контактной кислоты, и затем навеска окиси ртути (0,184 г), затем «утка» встряхивается в течение 5 минут для распределения катализатора в системе. Изолированная от внешней среды и герметичная система из газометра 3 промывается 1-1,5 л ацетилена на холоде! Только после этого к «утке» подключают нагрев. Температура в «утке» на протяжении всего опыта поддерживается постоянной равной 96 ° С; Для этого через внешний кожух «утки» продувается пар из паровика 4.
Перед началом опыта проводится нулевой отсчет объема газа в газометре и измерительной бюретке 5. Затем с помощью крана газометра и крана сифона бюретки устанавливается заданная скорость подачи ацетилена по градуированному реометру и включается мотор болтушки. При интенсивном встряхивании «утки» ацетилен с заданной скоростью пропускается через систему до тех пор, пока общий объем пропущенного ацетилена по газометру составит 4 л (каждом опыте). Непрореагировавший ацетилен вместе с продуктами реакции поступает из «утки» в шариковые приемники, наполненные дистиллированной водой. Ацетальдегид поглощается водой, а ацетилен проходит дальше в измерительные бюретки, позволяющие определить объем ацетилена, не вступившего в реакцию.
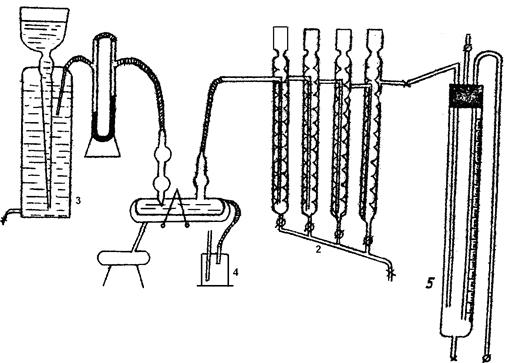
Рисунок 10.2 — Схема гидратации ацетилена по Кучерову
1 – «утка»; 2 – холодильник; 3 – газометр; 4 – паровик; 5 – бюретка
Для непрерывного проведения опыта система снабжена двумя измерительными бюретками. Как только одна бюретка заполнится газом, записывается объем газа в бюретке и ток ацетилена переключается на вторую бюретку. Первая бюретка соединяется с атмосферой и с помощью уравнительной «груши» ацетилен выбрасывается в атмосферу, бюретка заполняется насыщенным раствором поваренной соли и снова готова к измерению. Аналогичная операция повторяется со второй бюреткой. В ходе опыта смена бюреток осуществляется несколько раз, и каждый раз фиксируется объем ацетилена, собранный в бюретках.
Анализ водного раствора ацетальдегида производится по методике, предложенной Гуляевой. Определение основывается на способности ацетальдегида реагировать с солянокислым гидроксиламином и выделившаяся кислота оттитровывается 0,1 н раствором щелочи в присутствии смешанного индикатора метилоранж-индигокармин (1 часть 0,1 н раствора метилоранжа и одна часть 0,25 %-ого раствора индигокармина). Водный раствор гидроксиламина подвергается гидролизу и поэтому имеет собственную кислотность, которую необходимо определить. Для этого в коническую колбу наливается 10 мл дистиллированной воды и 5 мл 1н раствора гидроксиламина. После пятиминутного отстаивания смесь оттитровывается 0,1 н раствором щелочи в присутствии смешанного индикатора. Количество мл щелочи, ушедшей на титрование, определяется кислотностью гидроксиламина.
Анализ ацетальдегида ведется так: из мерной колбы на 1000 мл пипеткой 10 мл раствора переносятся в коническую колбу на 100 мл. Туда же вводится 5 мл 1н раствора гидроксиламина. Раствор стоит 5-8 минут, после чего к нему добавляется 3-5 капель смешанного индикатора и содержимое оттитровывается щелочью до зеленой окраски. (В кислой среде индикатор имеет фиолетовую окраску, а щелочной — зеленую).
Расчет проводится по формуле:
Г=(А-В)·0,0044·100
где Г — содержание ацетальдегида в граммах в 1л раствора;
А — количество мл 0,1 н раствора щелочи, пошедшей на титрование раствора;
В — кислотность гидроксиламина в мл ОД н раствора щелочи;
0,0044 — титр ацетальдегида в г, эквивалентный 1 мл 0,1 н раствора щелочи.
По данным титрования производится расчет выхода ацетальдегида в % на пропущенный и вступивший в реакцию ацетилен, в конце опыта записываются: объем пропущенного ацетилена (н.у.), выход ацетальдегида (%) на пропущенный и вступивший в реакцию ацетилен.
Контрольные вопросы
1. Реакция гидратации по Кучерову.
2. Способы получения и применение ацетальдегида.
3. Методика проведения работы.
4. Определение содержания ацетальдегида в растворе.
Литература
1. С.И.Вольфкович и др. Общая химическая технология, т. П, М., Госхимиз-дат, 1959 г., стр. 444.
2. Б.Н. Долгов. Катализ в органической химии. Л., Госхимиздат, 1959 г., стр.516.
§
Стирол относится к числу важнейших мономеров. Он производится в крупных масштабах и используется для получения разнообразных полимерных материалов: полистирола, синтетических каучуков, латексов и др.
Среди различных способов получения стирола в настоящее время в промышленности наибольшее распространение получил процесс каталитического дегидрирования этилбензола в присутствии паров воды:
С6Н5-СН2СН3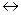 С6Н5-СН=СН2 Н2
С6Н5-СН=СН2 Н2
В качестве катализаторов используют железохромовые оксидные катализаторы, промотированные оксидом калия. Процесс проводят при 580 — 620 °С, мольном отношении этилбензола к воде, равном 1 : (17÷20), и объемной скорости подачи этилбензола 0,20 — 0,50 ч -1. При степени конверсии этилбензола 40 — 50% селективность процесса составляет около 90%.
Побочные продукты процесса (бензол, толуол, этилен, метан, смолистые вещества и др.) образуются в результате реакций термического распада, гидрогенолиза и уплотнения, как этилбензола, так и стирола:
Железохромовые оксидные катализаторы обладают способностью саморегенерации, так как они катализируют частично и реакцию водяного пара с коксом и смолистыми веществами, которые отлагаются на поверхности контакта:
С Н2О СО Н2
С 2Н2О C02 2H2
Селективность процесса дегидрирования на данном катализаторе и при данной температуре зависит от степени конверсии этилбензола. С увеличением степени конверсии и приближением ее к состоянию равновесия селективность процесса резко падает.
§
Реактивы
Этилбензол 112 г
Дистиллированная вода 800 мл
Катализатор КС-4 (или К-22), размер частиц 2X3 мм, 40 см3
Гидрохинон 3 г
Инертная насадка (кварц), размер частиц 2X3 мм, 25 см3
Дегидрирование этилбензола в стирол проводят на установке, схема которой приведена на рисунок 11. Установка состоит из реактора 1, двухсекционной электропечи 2, механического дозатора 6 для подачи этилбензола и воды, мерных цилиндров 7 для воды и этилбензола, переходника 8, милливольтметра 12, переключателя термопар 13, автотрансформаторов 14 и 15, кварцевого холодильника 16, ледяной бани 17, обратного холодильника 18 и приемника 19. Перед началом опытов проверяют правильность заполнения реактора катализатором и инертной насадкой, исправность термопар, надежность и герметичность всех соединений установки.
Реактор представляет собой кварцевую трубку с внутренним диаметром зоны испарения (I) и реакционной зоны (II), равным 18 мм, и суммарной длиной обеих зон 300 мм. Он снабжен четырьмя карманами 10 для термопар 11, выступами 5 для удерживания катализатора 4 и инертной насадки 3. Нижняя часть реактора после выступов сужена до 8 мм для уменьшения времени пребывания продуктов реакции в зоне высоких температур. Реактор заполняют катализатором и инертной насадкой так, как показано на рисунке .
Опыты проводят при двух температурах (570 и 600 °С), четырех объемных скоростях подачи этилбензола (0,2, 0,3, 0,4 и 0,6 ч»1) и постоянном мольном отношении этилбензола к воде, равном 1:18.
Градуируют дозаторы для подачи этилбензола и воды согласно заданным расходам и приступают к выполнению работы. Начинают опыт при температуре 570 °С и объемной скорости подачи этилбензола 0,2 ч -1. Для предотвращения преждевременного разрушения частиц катализатора и выхода его из строя в процессе работы строго придерживаются следующего графика нагрева и охлаждения реактора. Реактор нагревают от комнатной температуры до 400°С и охлаждают от 400 °С до комнатной температуры только под током азота. Азот подают в установку через кран 9 со скоростью 150 мл/мин. Выше 400°С до температуры опыта и от температуры опыта до 400 °С нагрев и охлаждение ведут при подаче в реактор воды со скоростью, которая была рассчитана для очередного опыта. Нагрев зоны испарения и реакционной зоны ведут одновременно со скоростью 5—6°С в минуту. Заданную скорость нагрева и температуру опыта регулируют с помощью автотрансформаторов 14 и 15. Температуру в зоне испарения в процессе опыта поддерживают на 3—5°С ниже, чем в реакционной зоне. Этилбензол начинают подавать в установку только после достижения и стабилизации заданной температуры в реакторе.
После 15 мин стабильной работы установки на данном режиме отбирают пробу реакционной массы для анализа. Пробу отбирают в течение 5 мин в пустой приемник 19. Органический слой отделяют от водного в делительной воронке, сушат над безводным хлоридом кальция и анализируют на хроматографе.
Для выполнения следующего опыта без остановки реактора вначале изменяют скорость подачи воды, а затем этилбензола и с помощью автотрансформаторов устанавливают необходимую температуру в обеих зонах реактора. Время и методика отбора пробы реакционной массы сохраняются такими же, как описано выше.
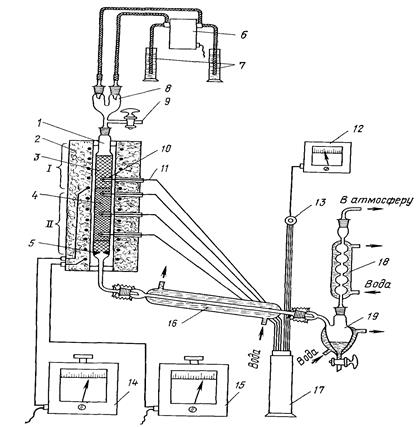
Рисунок 11 — Установка для дегидрирования этилбензола:
1 – реактор; 2 – двухсекционная печь; 3 – инертная насадка; 4 – катализатор; 5 – выступы; 6 – дозатор; 7 – мерные цилиндры; 8 – переходник; 9 – кран; 10 – карманы для термопар; 11 – термопары; 12 – милливольтметр; 13 — переключатель; 14, 15 – автотрансформаторы; 16, 18 – холодильники; 17 – ледяная баня; 19 – приемник.
На основании полученных данных для обеих температур строят графики зависимости: степень конверсии этилбензола — объемная скорость его подачи и селективность процесса — степень конверсии этилбензола.
Рассчитывают термодинамически возможную степень конверсии этилбензола в стирол для данных температур без учета образования побочных продуктов, используя уравнение для константы равновесия Кр:
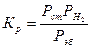
где Рст,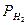 Рэб— парциальные давления стирола, водорода и этилбензола
Рэб— парциальные давления стирола, водорода и этилбензола
соответственно.
Для нахождения парциальных давлений веществ, участвующих в равновесии, вычисляют число молей каждого вещества в реакционной зоне в состоянии равновесия, принимая начальное число молей этилбензола за единицу:
Парциальные давления, выраженные через мольные доли веществ, участвующих в равновесии, и общее давление в системе будут следующими:
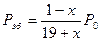
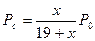
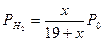
После подстановки этих выражений в начальное уравнение получают формулу
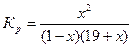
которую решают относительно х, и после несложных преобразований получают формулу для вычисления доли молей этилбензола, превращенной в стирол:
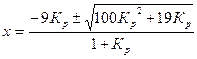
Для реакции дегидрирования этилбензола в стирол при температуре 580°С Кр=0,135, а при температуре 620°С КР=0,295.
Зная долю молей этилбензола х, превращенную в стирол, рассчитывают термодинамически возможную степень конверсии этил-бензола в стирол для данных температур:
X = х·100
На установке дегидрирования этилбензола по приведенной выше методике можно дегидрировать изопропилбензол и диэтилбензол. В случае дегидрирования изопропилбензола температуру реакции снижают на 20 °С, а при дегидрировании диэтилбензола уменьшают в два раза объемную скорость подачи углеводорода и мольное отношение диэтилбензола к воде.
Контрольные вопросы
1. Применение продуктов дегидрирования этилбензола.
2. Применяемые для процесса катализаторы.
3. Температура протекания процесса.
Литература
1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 142-147.
2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с.
§
Большое число важнейших химических соединений (циклогексан, циклогексанол, высшие спирты, амины, стирол, изобутилен, бутадиен-1,3 и др.) в промышленности получают при помощи процессов гидрирования и дегидрирования.
Реакции гидрирования и дегидрирования тесно связаны друг с другом, протекают в противоположных направлениях и в большинстве случаев находятся в термодинамическом равновесии:
АН2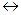 А Н2
А Н2
Реакция гидрирования сопровождается уменьшением объема реакционной массы и выделением тепла, а реакция дегидрирования — увеличением объема реакционной массы и поглощением тепла. Поэтому сдвиг равновесия в сторону гидрирования или дегидрирования зависит от условий процесса. Так, низкие температуры и высокие давления благоприятствуют реакции гидрирования, а высокие температуры и низкие давления — реакции дегидрирования.
Процессы гидрирования и дегидрирования, как правило, проводят в присутствии катализаторов, которые значительно повышают скорость процесса и его селективность. В качестве катализаторов применяют металлы и их сплавы (Pt, Pd, Ni, Cu, Zn и др.), оксиды металлов и их смеси (Fe2O3, Сr3О3,ZnO, MgO и др.), сульфиды металлов и их смеси (NiSi, WS3, MoS3 и др.).
Металлы являются наиболее активными катализаторами, поэтому их чаще всего используют в процессах гидрирования, которые проводят при температурах до 220 °С и давлении до 6 МПа. В процессах дегидрирования применяют преимущественно оксидные катализаторы, которые по сравнению с металлами хотя и менее активны, но при 500—650 °С обладают большей селективностью, стабильностью и легко регенерируются.
Циклогексан и циклогексанол являются важными промежуточными продуктами в процессе получения циклогексанона, капролактама и адипиновой кислоты. Кроме того, они широко используются как растворители.
В промышленности циклогексан и циклогексанол получают гидрированием бензола и фенола соответственно. Процесс проводят под давлением при мольном отношении гидрируемого вещества к водороду, равном 1: (10÷30). Например, бензол гидрируют на никелевом катализаторе при 180—200 °С и 1,5—2,0 МПа или на сульфидных катализаторах при л;300°С и 20—30 МПа:
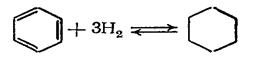
При гидрировании бензола на никелевом катализаторе повышение температуры выше 220 °С приводит к частичному распаду циклогексана на углерод, метан и другие побочные продукты и к. быстрой дезактивации катализатора.
Фенол гидрируют на никелевом катализаторе при температурах не выше 150°С и давлении 1,5—2,0 МПа в газовой фазе. При более высокой температуре значительная часть фенола гидрируется до циклогексанона и расходуется на образование других побочных продуктов:
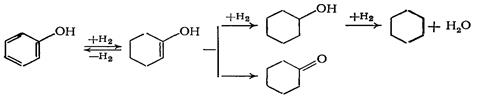
Циклогексанон можно гидрировать в циклогексанол, но условия гидрирования более жесткие. Селективность гидрирования бензола и фенола составляет 98% и выше.
§
Реактивы
Бензол, свободный от соединений серы 39 г
Водород (из баллона) 130 л
Катализатор — Ni/Сг2О3 (в таблетках) 100см3
Бензол гидрируют на установке, схема которой приведена на рисунке 12.
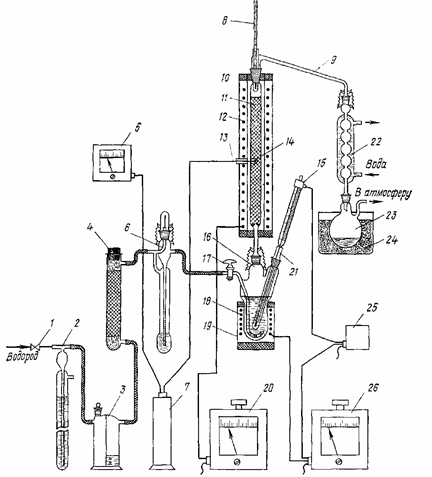
Рисунок 12 — Установка для гидрирования бензола в газовой фазе:
1 – игольчатый вентиль; 2 – гидравлический затвор; 3 – склянка Тищенко с концентрированной кислотой; 4 – трубка с хлоридом кальция безводным; 5 – милливольтметр; 6 – реометр; 7, 24 – ледяные бани; 8 – термометр; 9 — переходник с карманом; 10, 19 – предохранительный кожух; 11 – реактор; 12 — электропечь; 13 – термопара; 14 – карман для термопары; 15 – контактный термометр; 16 – испаритель; 17 – барботер с краном; 18 – электроспираль; 20, 26 — автотрансформаторы; 21 – съемный кран; 22 – обратный холодильник; 23 – приемная колба; 25 – реле-регулятор
Установка состоит из игольчатого вентиля 1, гидравлического затвора 2, склянки Тищенко 3, трубки с безводным хлоридом кальция 4, милливольтметра 5, реометра 6, ледяных бань 7 и 24, термометра 8, переходника с карманом 9, реактора 11, электроспиралей 12 и 18, термопары 13, контактного термометра 15, испарителя 16, автотрансформаторов 20 и 26, съемного кармана 21, обратного холодильника 22, приемной колбы 23 и реле-регулятора 25. Перед началом опыта проверяют правильность сборки, герметичность всех соединений установки и убеждаются в надежной работе системы автоматического поддержания в испарителе заданной температуры.
Реактор изготовлен из термостойкой стеклянной трубки внутренним диаметром 22 мм и длиной 300 мм, снабжен карманом 14 для термопары, выступами для удерживания катализатора и предохранительным кожухом 10. Испаритель 16 объемом реакционной зоны 60 мл также изготовлен из термостойкого стекла и снабжен барботером 17 для водорода, электроспиралью 18 для нагрева реакционной массы, съемным карманом 21 для контактного термометра и предохранительным кожухом 19.
Отсоединяют переходник 9 от обратного холодильника и присоединяют его через колбу (емкостью 100 мл) к линии сброса водорода в атмосферу. Подают водород в установку со скоростью 50 мл/мин и включают обогрев реактора.
По достижении температуры в реакторе 200—210 °С увеличивают скорость подачи водорода до 800 мл/мин и при этой температуре восстанавливают катализатор до прекращения выделения воды и очистки стенок переходника 9 от влаги. Во время восстановления катализатора готовят ледяную баню 7. После прекращения выделения воды уменьшают скорость подачи водорода до 50 мл/мин, переходник 9 снова присоединяют к обратному холодильнику, а линию сброса водорода — к приемной колбе 23.
Условия проведения опыта следующие: температура в реакторе 200—210 °С, температура в испарителе 35 °С (при гидрировании фенола в реакторе 148—160°С, в испарителе 135°С), скорость подачи водорода 800 мл/мин.
Через горло для съемного кармана 21 наливают в испаритель 39 г бензола и включают обогрев испарителя. По достижении в испарителе заданной температуры (35°С) увеличивают скорость подачи водорода до 800 мл/мин. Момент увеличения скорости подачи водорода принимают за начало опыта. Гидрирование ведут до полного испарения бензола из испарителя и прекращения конденсации циклогексана в холодильнике. Затем уменьшают скорость подачи водорода до 50 мл/мин, выключают обогрев реактора и испарителя. Момент уменьшения скорости подачи водорода принимают за конец опыта. После охлаждения реактора до комнатной температуры прекращают подачу водорода, закрывают кран на барботере и выход из реактора после переходника 9. Вход и выход из реактора должны быть плотно закрыты, чтобы в него не мог проникнуть воздух.
Продукты реакции из колбы 23 переносят в предварительно взвешенную на технических весах колбу, определяют массу и показатель преломления. Зная показатель преломления чистого бензола и циклогексана и исходя из того, что для показателя преломления в смеси сохраняется правило аддитивности, вычисляют концентрацию образовавшегося циклогексана С, степень конверсии бензола X и потери У (в % масс.):
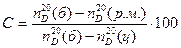

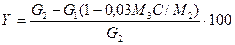
где  (б), (ц), (р.м.) — показатели преломления бензола, циклогексана и реакционной массы соответственно; G2, G1 — масса поданного на реакцию бензола и реакционной смеси соответственно, г; М1, М2, М3 — молекулярные массы бензола, циклогексана и водорода соответственно.
(б), (ц), (р.м.) — показатели преломления бензола, циклогексана и реакционной массы соответственно; G2, G1 — масса поданного на реакцию бензола и реакционной смеси соответственно, г; М1, М2, М3 — молекулярные массы бензола, циклогексана и водорода соответственно.
Рассчитывают объем пропущенного водорода и степень его полезного использования.
Контрольные вопросы
1. Влияние катализаторов на протекание реакций гидрирования.
2. Температурный режим процесса.
3. Как определяют показатель преломления чистого бензола?
Литература
1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 138-142.
2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с.
§
Реактивы
Изопропилбензол 90,2 г (105 мл)
Гидропероксид изопропилбензола 2 г
Воздух или технический кислород (из баллона)
Уксусная кислота ледяная 200 мл
Иодид калия, 50%-ный водный раствор 50 мл
Тиосульфат натрия, 0,1 н. водный раствор 700 мл
Крахмал, водный раствор 40 мл
Изопропилбензол окисляют в гидропероксид на установке, схема которой приведена на рисунке 13. Установка состоит из игольчатого вентиля 1 для регулирования подачи сжатого воздуха, трубки 2 с активированным углем, трубки 3 сбезводным хлоридом кальция, реометра 4 для измерения скорости подачи воздуха, предохранительной склянки 5, реактора 8, контактного термометра 9, обратного холодильника 10, сепаратора 11, реле-регулятора 14 и автотрансформатора 15. Перед началом опыта проверяют правильность сборки, герметичность всех соединений установки и надежность работы системы для поддержания в реакторе заданной температуры.
Реактор изготовлен из термостойкого стекла (d=24 мм, h = 250 мм) и снабжен барботером 7 для подачи воздуха, отводной трубкой с краном 13 для отбора проб, электроспиралью 6 для нагрева реакционной массы.
В сухой чистый реактор наливают 100 мл 3%-ного изопропилбензольного раствора гидропероксида изопропилбензола (или 100 мл изопропилбензола), 0,03 г стеарата кобальта и 0,03 г гидроксида кальция. Подают воду в обратный холодильник и включают обогрев реактора. По достижении в реакторе заданной температуры 115±0,5°С и стабилизации ее включают подачу воздуха со скоростью 800 мл/мин. Момент включения подачи воздуха принимают за начало опыта.
В ходе опыта каждые 30 мин в колбы емкостью 100 мл с притертыми пробками, предварительно взвешенные на аналитических весах, отбирают две параллельные пробы по 1 мл для определения концентрации гидропероксида изопропилбензола в реакционной массе. Перед отбором проб отводную трубку с краном 13 продувают воздухом при помощи резиновой груши.
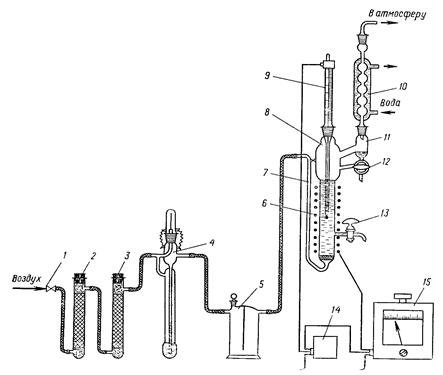
Рисунок 13 — Установка для окисления изопропилбензола в гидропероксид изопропилбензола:
1— игольчатый вентиль; 2 — трубка с активированным углем; 3 — трубка с безводным хлоридом кальция; 4 — реометр; 5 — предохранительная склянка; 6 — электроспираль; 7 — барботер; 8 — реактор; 9 — контактный термометр; 10 — обратный холодильник; 11 — сепаратор; 12 — трехходовой кран; 13 — кран; 14 — реле-регулятор; 15 — автотрансформатор.
Окисление изопропилбензола заканчивают по достижении концентрации гидропероксида в реакционной массе, ≈20% (масс.). Продолжительность опыта 3—4 ч. По окончании опыта выключают подачу воздуха, обогрев реактора и сразу отсоединяют линию воздуха от реактора для предотвращения засасывания в нее реакционной массы.
Реакционную массу после охлаждения выливают в предварительно взвешенную колбу емкостью 150 мл, определяют массу, состав и составляют материальный баланс опыта.
Массу продуктов реакции G1 в пересчете на изопропилбензол вычисляют по формуле:
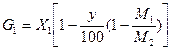
Общую массу G2 взятых на анализ проб (gi) в пересчете на изопропилбензол вычисляют по формуле:
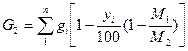
Потери изопропилбензола рассчитывают по формуле:
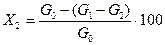
где М1, M2— молекулярные массы изопропилбензола и гидропероксида изопропилбензола; gi— масса i-той пробы, г; yi— концентрация гидропероксида изопропилбензола в i-той пробе, % (масс.); п — число проб, взятых на анализ.
На основании полученных данных строят график зависимости: концентрация гидропероксида изопропилбензола (в % масс.) — продолжительность реакции (в мин); формулируют свои выводы о выполненной работе.
Контрольные вопросы
1. Применение фенола и ацетона в промышленности.
2. Стадии производства фенола и ацетона.
3. Формула по определению массы продуктов реакции.
4. Для чего в лабораторную установку включена трубка с безводным хлоридом кальция?
Литература
1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 111-119.
2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с.
§
Реактивы
Сжатый воздух
Этилен (из баллона) 60—70 л
Катализаторный раствор 200 мл
Гидроксиламин солянокислый, 0,5 н. водный раствор 500 мл
Гидроксид натрия, 0,1 н. водный раствор 700 мл
Индикатор — 0,1%-ный бромфеноловый синий 3 мл
Окисление этилена в ацетальдегид проводят на установке, схема которой приведена на рисунке 14. Установка состоит из игольчатых вентилей 1 и 2, реометров 3, предохранительных склянок 4, двухсекционного реактора 7, снабженного электроcпиралями 5, барботерами 6, контактными термометрами 8, переливными трубками 10 и эффективными обратными холодильниками 9, реле-регуляторов 11, автотрансформаторов 12, капельных воронок 13, тройника 14, абсорберов 15, снабженных рубашками для охлаждения и стеклянными насадками 16, и приемников 17.
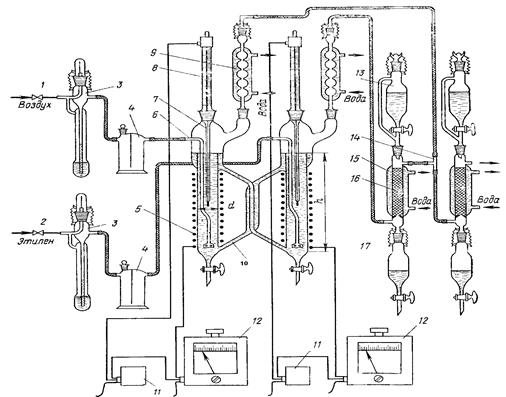
Рисунок 14 — Установка для окисления этилена в ацетальдегид:
1, 2— игольчатые вентили; 3 — реометры; 4 — предохранительные склянки; 5 — электроспирали; 6 — барботеры; 7 — реактор; 8 — контактные термометры; 9 — обратные холодильники; 10 — переливные трубки;
11— реле-регуляторы; 12 — автотрансформаторы; 13 — капельные воронки; 14 — тройник; 15 — абсорберы; 16 — стеклянная насадка; 17 — приемник; I— секция для регенерации катализатора; II — секция для окисления этилена.
Перед началом опыта тщательно проверяют правильность сборки, герметичность всех соединений установки и убеждаются в надежной работе систем. Для регулирования и поддержания температуры в обеих секциях реактора. Реактор, изготовленный из термостойкого стекла, состоит из двух секций с объемом по 100 мл, диаметром 24 мм и высотой 250 мм; I секция — для регенерации катализатора, II секция — для окисления этилена.
Абсорберы также изготовлены из термостойкого стекла; часть абсорберов, заполненная стеклянной насадкой, имеет объем 60 мл, диаметр 20 мм и высоту 200 мм.
Градуируют капельные воронки для подачи воды в абсорберы и только после это приступают к выполнению работы.
Проводят две серии опытов. В первой серии при постоянной температуре в обеих секциях реактора (85°С) и постоянной скорости подачи воздуха (60 мл/мин) изменяют скорость подачи этилена (130, 150, 170 и 190 мл/мин). Во второй серии при постоянной температуре в обеих секциях реактора (85°С) и постоянной объемной скорости подачи этилена (170 мл/мин) изменяют скорость подачи воздуха (100, 80, 60 и 40 мл/мин). Продолжительность каждого опыта 80 мин.
Наливают в реактор 200 мл предварительно приготовленного катализаторного раствора), включают подачу воды в обратные холодильники и в рубашки абсорберов, наливают воду в капельные воронки, включают подачу воздуха в первую секцию реактора со скоростью 20 мл/мин и обогрев обеих секций реактора. По достижении в обеих секциях реактора температуры 85±0,5°С включают подачу воды из капельных воронок в абсорберы со скоростью 6 мл/мин, подачу этилена во вторую секцию реактора и увеличивают скорость подачи воздуха в первую секцию реактора до заданной. Подают газы плавно, чтобы избежать переброса или перетока катализаторного раствора. Момент включения подачи этилена принимают за начало опыта.
В ходе опыта непрерывно наблюдают за исправной работой всех систем установки, поддерживают параметры опыта на заданном уровне. Периодически пополняют запасы воды в капельных воронках и выгружают раствор ацетальдегида из приемников.
Реакционные газы из второй секции реактора проходят через обратный холодильник 9, первый абсорбер 15, где поглощается основное количество ацетальдегида, смешиваются в тройнике 14 с газами первой секции реактора, проходят через второй абсорбер и выводятся под тягу.
По окончании опыта выключают подачу этилена и подачу воды в первый абсорбер, а подачу воздуха продолжают еще в течение 5 мин. Затем выключают подачу воздуха и подачу воды во второй абсорбер.
Водные растворы ацетальдегида из первого и второго абсорберов объединяют, определяют их общий объем. Этот раствор анализируют на содержание ацетальдегида по методике.
Перед началом следующего опыта и в конце работы проверяют уровень и рН катализаторного раствора в реакторе. Если уровень катализаторного раствора понизился, его восстанавливают, добавляя дистиллированную воду. Если рН катализаторного раствора окажется больше двух, нему добавляют несколько капель концентрированной соляной кислоты и при 85°С через раствор пропускают воздух со скоростью 20 мл/мин в течение 1 ч.
По окончании работы и охлаждения реактора катализаторный раствор сливают в сосуд с притертой пробкой и хранят в темном месте. Если катализаторный раствор потерял свою активность, его заменяют свежим, который готовят по методике, описанной ниже, а из отработанного катализаторного раствора выделяют палладий.
На основании данных, полученных в результате выполненной работы, строят графики зависимости: выход ацетальдегида — объемная скорость подачи этилена и воздуха. Формулируют свои выводы о проведенной работе.
§
В отдельных колбах готовят два раствора. В колбе емкостью 500 мл в 300 мл дистиллированной воды растворяют при перемешивании 3 г хлорида железа и 50 г хлорида меди (СuСl2). После растворения солей раствору дают отстояться в течение суток. В другой колбе емкостью 200 мл в 20 мл 10%-ной соляной кислоты и 25 мл уксусной кислоты растворяют 2 г хлорида палладия и этот раствор приливают к первому. Раствор хлорида палладия готовят перед смешением.
Полученный раствор переливают в мерный цилиндр емкостью 500 мл и доводят его объем дистиллированной водой до 400 мл, сливают в колбу с притертой пробкой и хранят в темном месте.
Катализаторный раствор используют только после выдержки не менее 12 ч и определения рН перед заполнением реактора, который должен быть не более 2. Если рН раствора>2, то, добавляя к нему по каплям концентрированную соляную кислоту, доводят его рН≈1,5.
Катализаторный раствор в темном месте в колбе с притертой пробкой может храниться продолжительное время практически без изменения активности.
Выделение палладия из отработанного катализаторного раствора
В процессе работы катализаторный раствор постепенно теряет свою активность вследствие накопления в нем высококипящих органических соединений. Поэтому примерно после 80 ч работы возникает необходимость замены катализаторного раствора свежим. Для выделения палладия из отработанного катализаторного раствора кислый Катализаторный раствор разбавляют равным объемом дистиллированной воды и приливают к нему водно-спиртовой раствор диметилглиоксима
СН3С-С-СН3
|| ||
НОN NОН
Диметилглиоксим образует с палладием нерастворимый в реакционной массе комплекс, который выпадает в осадок. После проверки полноты осаждения палладия осадок отделяют на воронке Бюхнера при отсасывании водоструйным насосом, промывают дистиллированной водой и сушат в вакуум-сушильном шкафу. Затем сухой осадок загружают в фарфоровый тигель и прокаливают в муфельной печи при 900±10°С в течение 1 ч. В результате такой обработки выделяется мелкодисперсный металлический палладий, который можно вновь использовать для получения хлорида палладия.
Контрольные вопросы
1. Роль хлорида палладия в данном процессе.
2. Технологические параметры проведения процесса.
3. Методика приготовления катализаторного раствора.
Литература
1. Одабашян Г.В. Лабораторный практикум по химии ТООНХС. М., Химия, 1982, с. 106-110.
2. Воскресенский П.И. Техника лабораторных работ. 10-е изд. М.. Химия, 1973, 717 с.
Лабораторная работа №15
Тема: Получение компаундированного дорожного битума на основе
Асфальта деасфальтизации
Теоретические основы
Дорожные битумы являются вяжущим для получения дорожных покрытий (асфальтобетона). Битумы по своему химическому составу состоят из масляной или углеводородной части и неуглеводородной части (смолы, асфальтены, карбены и корбоиды), являются высоковязкими продуктами процессов переработки нефтяных остатков. Битумы по способу получения бывают остаточные (продукты глубоковакуумной перегонки мазутов), окисленные (продукты окисления гудронов кислородом воздуха) и компаундированные (полученные смешением).
С целью получения битумов, различающихся по эксплуатационным свойствам, и использования остатков деасфальтизации применяют метод компаундирования асфальта деасфальтизации и продуктов перегонки нефти: мазутов и гудронов. Компаундированные битумы отличаются от окисленных высокой растяжимостью при 25 °С, низкой температурой хрупкости, достаточно широким диапазоном пластичности при температуре эксплуатации.
Цель работы
Получить дорожный битум методом компаундирования, освоить основные методы определения качества дорожных битумов.
Материалы и оборудование
1. Сырьё – гудрон, мазут, асфальт деасфальтизации.
2. Электроплитка.
3. Металлические пенетрационные чашки ёмкостью 100 ÷ 150 г.
4. Пенетрометр.
5. Прибор для определения температуры размягчения.
6. Водяная баня.
7. Электромешалка.