

Дополнение. гаусманнит и другие смешанные оксиды марганца
Смешанный оксид Mn3O4 содержит марганец в степенях окисления 2 и 4. Он встречается в виде минерала гаусманнита, образующего черные с металлическим блеском кристаллы. При измельчении они превращаются в черно-красный порошок. Оксид Mn3O4 образуется при нагревании до 1000 оС любых соединений марганца на воздухе или в кислороде. При этом он кристаллизуется в тетрагональной низкотемпературной модификации α-Mn3O4, идентичной природному гаусманниту. Она имеет структуру, близкую шпинели. Выше 1170 оС гаусманнит превращается в высокотемпературную кубическую β-форму со структурой нормальной шпинели. В ней ионы Mn3 занимают только октаэдрические позиции. Действительно, для ионов с конфигурацией d4 характерна большая энергия стабилизации октаэдрическим окружением, 95 кДж/моль, показывающая предпочтение октаэдрических позиций по сравнению с тетраэдрическими. Это значение ЭСКП уступает только иону Cr3 (158 кДж/моль). Поэтому все шпинели трехвалентного марганца относятся к нормальным шпинелям.
Mn3O4 образуется при термическом распаде диоксида, который при 550 оС переходит в Mn2O3, а при 950 – в Mn3O4. Термический распад сульфата MnSO4 выше 1000 оС или сплавление сульфата марганца(II) c поташом дают гаусманнит в виде красных кристаллов. Чистый Mn3O4 получают разложением карбоната марганца(II) в инертной атмосфере:
3MnСО3 = Mn3O4 СO↑ 2СО2↑.
Это вещество является также конечным продуктом при окислении гидроксида марганца(II) на воздухе.
Многие восстановители, например, водород и углерод, способны переводить его лишь в низший оксид MnO. Полное восстановление оксида марганца (II, IV) происходит только в присутствии железа, которое растворяет марганец.
При нагревании на воздухе гаусманнит окисляется до оксида марганца(III), а действием хлора на водную суспензию Mn3O4 удается получить оксид марганца(IV).
Восстановление гауманнита сернистым газом в газовой фазе и в растворе приводит к различным продуктам:
Mn3O4 SO2 = 2MnO MnSO4,
Mn3O4 2SO2 2H2SO4= 2MnSO4 MnS2O6 H2O.
В водных растворах кислот происходит диспропорционирование:
Mn3O4 4Н = MnO2 2Mn2 2Н2О.
Благодаря наличию большого числа неспаренных электронов, гаусманнит обладает необычными магнитными свойствами. При изменении температуры он испытывает ряд магнитных переходов с упорядочением электронных спинов.
Интересно, что оксид Mn3O4 оказывает более слабое каталитическое действие на разложение пероксида водорода, чем оксиды марганца(II) и марганца(IV).
В системе марганец-кислород есть также промежуточный оксид Mn5O8, образующийся в виде тонких блестящих желтых пластинок при нагревании MnOОН в токе кислорода до 250 оС или из MnO в гидротермальных условиях при повышенном давлении кислорода. На основании изоструктурности с Cd2Mn3O8 ему приписана формула Mn2IIMn3IVO8, в его решетке ионы Mn4 расположены в искаженных октаэдрах из атомов О на расстояниях 0,185-0,192 нм. Ионы Mn2 находятся в искаженных тригональных призмах с тремя короткими, 0,213 нм, и тремя длинными, 0,230 нм, связями Mn-O (рис.5.14.Структура оксида Mn5O8). При нагревании на воздухе или в кислороде Mn5O8 переходит в α-Mn2O3.
КОНЕЦ ДОПОЛНЕНИЯ.
Низшие оксиды технеция и рения со степенями окисления ниже 4 неустойчивы и мало изучены. Диоксиды ЭО2 известны для всех трех металлов седьмой группы: у марганца это наиболее часто встречающийся в природе минерал пиролюзит. Красно-коричневый ТсО2 — самый устойчивый оксид технеция: он является конечным продуктом высокотемпературного разложения многих кислородных соединений. Сине-черный диоксид рения, несмотря на многие общие свойства с оксидом технеция(IV), при высоких температурах подвергается диспропорционированию на Re2O7 и металл.
К настоящему времени известно не менее четырнадцати соединений состава MnO2, представляющие собой порошки черно-коричневого цвета. Формально они считаются полиморфными модификациями диоксида, хотя их составы часто существенно отличаются от стехиометрического. Многие из этих фаз дефицитны по кислороду MnOх (1,7<x<2), часто содержат воду, ионы ОН–, катионы металлов, часть ионов Mn4 в них может быть замещена на Mn2 . Например, в решетку α-MnO2, образовавшегося при окислении сульфата марганца(II) раствором персульфата аммония, встроены ионы NH4 , в ней содержится 91,5 % MnO2. Известны три модификации диоксида марганца стехиметрического состава – пиролюзит (β-MnO2), имеющий тетрагональную структуру типа рутила (Сноска: Известен также и пиролюзит нестехиометрического состава MnOх, х = 1,93 –2,00), рамсделлит, и λ-MnO2. Все они образованы октаэдрами MnO6, связанными в цепи или слои (Рис.5.15. Кристаллические структуры рамсделлита (а) и пиролюзита (б)). γ-Модификация диоксида, используемая для производства гальванических элементов, имеет состав MnO1,93. Она состоит из фрагментов структур рамсделлита и пиролюзита, и содержит большое число различных дефектов (J.-R. Hill, C.M. Freeman, M.H. Rossouw, J. Solid State Chem., 2004, 177, 165).
Среди многочисленных реакций образования MnO2 только некоторые предпочтительны, благодаря легкости проведения и воспроизводимости. К ним относятся окисление солей соединений марганца(II) растворами хлоратов, перманганатов, персульфатов, хлором и озоном
3Mn(OH)2 NaClO3 = 3MnO2 NaCl 3H2O,
3MnCl2 2KMnO4 2H2O = 5MnO2 4HCl 2KCl,
восстановление перманганатов действием пероксида водорода, соляной кислоты, сернистого газа, сульфитов, спиртов, солями марганца(II):
2KMnO4 3SO2 2H2O = 2MnO2 2KHSO4 H2SO4
4KMnO4 3C2H5OH = 4MnO2 KOH 3CH3COOK 4H2O.
Большая часть диоксида марганца выделяется в виде бурого осадка, однако, часть его остается в виде коллоидного раствора.
Описанные выше способы приводят к продукту, представляющему собой смесь различных модификаций MnO2. Для получения той или иной модификации в чситом виде используют особые методы синтеза. При термическим разложении нитрата Mn(NO3)2. ниже 350 °C образуется γ-модификация, при более высокой температуре — β-MnO2. При выдерживании вещества в токе О2 при 400-500 оС удается получить продукт стехиометрического состава. При электролитическом окислении растворов солей образуется нестехиометрическая γ-модификация, в продуктах сопропорционирования солей марганца(II) с перманганатами всегда присутствуют ионы Mn2 . При окислении соединений марганца(II) пероксидом водорода, хлором, бромом, гипогалогенатами, хлоратами, броматами, персульфатами образуется диоксид MnО2-х с дефицитом кислорода.
Получение пиролюзита восстановлением перманганата пероксидом водорода проводят в слабо кислом, нейтральном или щелочном растворе. При этом пероксид водорода необходимо брать в избытке, учитывая его каталитическое разложение образующимся пиролюзитом. В сильно кислой среде пероксид водорода восстанавливает пиролюзит до солей марганца(II):
MnO2 H2O2 2H = Mn2 2H2O O2↑.
Диоксид марганца, как соединение промежуточной степени окисления, проявляет окислительные и восстановительные свойства. Для него более характерно поведение окислителя. При нагревании происходит внутримолекулярная окислительно-восстановительная реакция, сопровождающаяся выделением кислорода и постепенным понижением степени окисления марганца с последовательным образованием различных оксидов:
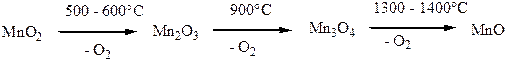
Процессы термического распада различных модификаций диоксида MnО2 протекают сложно и зависят от стехиометрии данного образца, его термической предыстории, а также от присутствия воды и других примесей, парциального давления О2, размера частиц. При постепенном нагревании MnО2 сначала отдает воду, полное обезвоживание достигается только при 600 оС. Одновременно в интервале 300-450 оС протекают полиморфные переходы между различными фазами MnО2 (например, γ-MnО2 превращается в β-MnО2 при 280-420 оС). При 500 – 600 оС MnО2 отщепляет кислород и переходит в Mn2О3, при 900 оС образуется Mn3О4 и О2, а при 1300 – 1400 оС – MnО. В присутствии кислорода разложение диоксида марганца происходит при большей температуре. При 1540 оС парциальное давление О2 над Mn3О4 достигает 0,1 атм.
Водородом диоксид восстанавливается до MnО, активные металлы, например, кальций или алюминий, а также ионные гидриды, восстанавливают его до металла:
3MnO2 4Al = 2Al2O3 3Mn, ΔH = — 1090 кДж/моль.
MnО2 СаН2 = Mn СаО Н2О.
С сухим сероводородом реакция протекает очень быстро уже при комнатной температуре:
MnО2 2H2S = MnS S 2H2O.
При пропускании H2S через водную суспензию MnО2 образуется белесый осадок, состоящий из смеси сульфида марганца(II) и серы; в растворе присутствуют ионы SO42- и S2O32-.
Выше 300 оС тонко размельченный порошок MnO2 взаимодействует с сернистым газом: MnO2 SO2 = MnSO4. В присутствии воды, наряду с сульфатом MnSO4, образуется дитионат MnS2O6 – это лабораторный метод синтеза дитионатов. Количество дитионата определяется модификацией диоксида: в случае β-MnО2 его выход достигает 97 %, а с α-MnО2 – 79 %. Тиосульфат в растворе при рН 4-5,8 окисляется пиролюзитом в сульфат и политионат:
3S2O32- 4MnO2 6H = 2SO42- S4O62- 4Mn2 3H2O.
Окислительные свойства диоксида марганца сильнее всего проявляются в сильно кислой среде, что следует из уравнения Нернста:
MnO2 4H 2 e– ® Mn2 2H2O, E0 = 1.23 B
E(MnO2/Mn2 ) = 1.23 – 0.118pH.
В кислой среде пиролюзит восстанавливается гидразином до Mn2 :
2MnO2 N2H4.H2SO4 H2SO4 = 2MnSO4 4H2O N2↑,
а в аммиачном растворе — лишь до Mn3 :
4MnO2 N2H4.H2SO4 2NH3 = 4MnOOH (NH4)2SO4 N2↑.
Бромиды и иодиды в кислой среде восстанавливают оксид марганца(IV) в соли марганца(II), окисляясь до простых веществ:
Реакция с хлорид-ионами протекает лишь в сильно кислых концентрированных растворах при нагревании, например, при взаимодействии с концентрированной соляной кислотой:
80 °C
MnO2 4HCl = MnCl2 Cl2↑ 2H2O,
или хлоридом, подкисленным азотной кислотой:
100 оС
2NaCl 4HNO3 MnO2 = 2NaNO3 Cl2↑ Mn(NO3)2 2H2O.
При обработке диоксида марганца холодной концентрированной HCl наблюдается образование темного раствора, содержащего молекулы тетрахлорида MnCl4 – его образование зафиксировано при температуре ниже 0 оС. Постепенно раствор светлеет вследствие распада тетрахлорида на хлор и MnCl2.
В плавиковой кислоте диоксид марганца восстанавливается до трифторида:
4MnO2 12HF = 4MnF3 O2 6H2O.
В азотной и разбавленной серной кислотах MnO2 не растворим, лишь при длительном хранении его под раствором кислоты наблюдается постепенное выделение кислорода вследствие окисления воды. Горячая концентрированная серная кислота также реагирует с MnO2, окисляясь до кислорода:
2MnO2 4Н = 2 Mn2 2Н2О О2↑.
Реакция протекает через стадию образования Mn3 , о чем свидетельствует окрашивание раствора в фиолетовый цвет. Образованием этих ионов также объясняется и быстрое растворение MnO2 в серной кислоте при добавлении соли марганца(II):
MnO2 Mn2 4H = 2Mn3 2H2O.
Взаимодействие диоксида MnO2 со щелочами протекает лишь в концентрированных растворах или расплавах. В зависимости от температуры реакции, соотношения реагентов, наличия или отсутствия окислителей, например, кислорода воздуха, образуются анионные оксоманаганаты, содержащие марганец в различных степенях окисления от 3 до 6. Например, при взаимодействии диоксида марганца с горячим концентрированным раствором гидроксида калия в инертной атмосфере сначала образуется зеленый раствор манганата(IV), который затем переходит в темно-синий раствор, содержащий эквимолярные количества манганата(V) и манганата(III). При проведении реакции на воздухе образуется манганат(VI) К2MnO4
2MnO2 4KOH O2 = 2K2MnO4 2H2O,
а в токе О2 при 600-800 оС – манганат(V):
2MnO2 6KOH 1/2O2 = 2K3MnO4 3H2O.
При атмосферном давлении воздуха реакция получения K2MnO4 начинается при 170 оС. При повышении давления до 10-19 кбар она ускоряется, и выше 19 кбар происходит взрыв. В отсутствии КОН манганат(VI) не образуется. При мольном отношении Mn:К = 4:1 на воздухе выше 450 оС образуется K2Mn8O16, при отношениях от 4:1 до 2:1 — K2Mn4O8, при еще меньшем отношении 1:3 уже при 250 оС получается K2MnO4. Для получения этой соли вместо кислорода в щелочном расплаве используют хлорат KClO3 или нитрат калия KNO3.
При добавлении диоксида марганца и перманганата калия в расплавленную щелочь образуется сине-зеленый расплав:
MnO4— MnO2 4ОН— MnO42- MnO43- 2Н2О.
MnO42- MnO43- 2Н2О.
При высоких концентрациях щелочи равновесие сдвигается вправо: выпадают синие кристаллы манганата Na3MnO4.xH2O, а манганат(VI) остается в растворе.
При окислении расплавом нитрита натрия NaNO2 диоксид марганца сначала образует Na3MnO4 и затем Na2MnO4. При нагревании смеси порошка MnO2 с пероксидами щелочных и щелочноземельных металлов образуются манганаты(VI):
MnO2 К2О2 = К2MnO4.
Действием еще более сильных окислителей, например, хлора в сильно щелочных растворах, MnO2 сразу переводится в перманганат.
Окисление атомов марганца в пиролюзите до высшей степени окисления реализуется также при кипячении с раствором персульфата аммония:
2MnO2 3(NH4)2S2O8 4H2O = 2HMnO4 3(NH4)2SO4 3H2SO4.
В этих реакциях диоксид марганца выступает в роли восстановителя.
Реакционная способность диокида марганца в реакциях ионного обмена намного ниже его окислительно-восстановительной активности, что является следствием его инертности – она свойственна всем ионам с конфигурацией d3. Именно поэтому, несмотря на амфотерный характер, это вещество в мягких условиях не реагирует с растворами кислот и щелочей, если при этом невозможны окислительно-восстановительные процессы. Об амфотерности диоксида марганца свидетельствует факт существования солей кислородсодержащих кислот, например, Mn(SO4)2, а также анионных манганатов(IV). Последние могут быть получены при нагревании пиролюзита с оксидами активных металлов. Например, взаимодействие c MgO приводит к Mg2MnO4 и Mg6MnO8, а c CaO – к CaMnO3, Ca2MnO4 и CaMn3O7.
Дополнение. Элемент Лекланше.
Ежегодно мировая промышленность производит около полмиллиона тонн диоксида марганца, большая часть которого идет на производство сухих электрохимических батарей. Из них наиболее обычны переносные сухие углерод-цинковые гальванические элементы, впервые предложенные французским физиком Ж. Лекланше в 1865 г.. Схему элемента можно схематично представить в виде
С│ MnO2 │≈10 % NH4Cl│Zn.
Сухой элемент Лекланше состоит из цинковой банки с вставленным в нее угольным электродом, погруженным в смесь диоксида марганца и графита. Цинковая банка помещена в крахмальную пасту, содержащую ZnCl2 и избыток твердого NH4Cl. Углерод выполняет роль положительного электрода – катода, цинк служит анодом, а диоксид марганца вводится в качестве окислителя, препятствующего выделению водорода на углероде. В настоящее время используют электролит, содержащий 8,8 % ZnCl2, 26 % NH4Cl,65,2 % H2O и небольшое количество сулемы HgCl2, играющей роль ингибитора. В ходе работы элемента происходит восстановление MnO2 и растворение цинкового анода:
Анод Zn 2NH3 – 2e– ® Zn(NH3)22 ,
Катод 2MnO2 2NH4 2e– ® 2MnOOH 2NH3
(сноска: Zn(NH3)22 — усредненная формула смеси комплексных аммиакатов и основных солей цинка разного состава. Продуктами восстановления диоксида марганца, помимо MnOOH, являются MnCl2 и ZnMn2O4). Особое значение MnO2 обусловлено его высокой окислительной способностью и практической нерастворимостью в электролите, благодаря чему вторичные реакции сводятся к минимуму.
Элемент обеспечивает напряжение порядка 1,6 В. В начале процесса значение рН составляет около 5 и во время разрядки батареи возрастает до 8-9.
Для использования в элементах Лекланше пригодны только определенные типы диоксида. Непосредственно для этой цели может быть использован только природный пиролюзит высшего качества, поэтому предпочтительнее употреблять синтетический γ-MnO2 получаемый электрохимически. К преимуществам сухого элемента Лекланше относится относительное длинное время работы, к недостаткам – высокое электрическое сопротивление и непостоянство потенциала. Создание новых ячеек с катодом MnO2 и модифицированным анодом, содержащим магний вместо цинка, обеспечивает высокое рабочее напряжение.
Рис. 5.16. Внешний вид (а) и устройство (б) элемента Лекланше.
КОНЕЦ ДОПОЛНЕНИЯ
Диоксиды технеция и рения представляют собой черные порошки, нерастворимые в воде. Диоксид технеция имеет искаженную структуру рутила, а диоксид рения кристаллизуется в моноклинной (низкотемпературный α-ReO2, структурный тип МоО2) или ромбической (высокотемпературный β-ReO2) структуре. Переход α-ReO2 ® β-ReO2 происходит необратимо при 400-650 оС.
Наиболее чистые препараты получены термическим разложением пертехнетата и перрената аммония в инертной атмосфере (700-750 и 300-450 оС, соответственно) или в
вакууме:
2NH4ReO4 = 2ReO2 N2↑ 4H2O.
Высшие оксиды восстанавливаются металлическим рением до диоксида:
550 оС
2Re2O7 3Re = 7ReO2.
Выше 700 оС реакция протекает в обратном направлении. Оксид технеция(IV) при высокой температуре возгоняется, не претерпевая разложения вплоть до 1100 оС. Таким образом, термическая устойчивость диоксидов возрастает в ряду MnO2 – ReO2 – TcO2.
В отличие от пиролюзита, для оксидов технеция(IV) и рения(IV) окислительные свойства не характерны. Например, при их растворении в концентрированной соляной кислоте образуются хлорокомплексы:
ReO2 6HCl = H2[ReCl6] 2H2O.
Действием концентрированной HNO3, пероксида водорода, бромной воды анионы гексахлорорената [ReCl6]2- окисляются до рениевой кислоты HReO4.
В реакциях диоксиды, как правило, выступают в роли восстановителей. Так, при нагревании на воздухе они не разлагаются, подобно MnO2, а наоборот, окисляются до высших оксидов М2О7. Диоксид технеция реагирует с галогенами, образуя оксогалогениды. Оба оксида восстанавливаются водородом до простых веществ (TcO2 при 500 оС, ReO2 – при 300 оС).
Диоксиды технеция и рения – амфотерные оксиды со слабо выраженными основными и кислотными функциями. Они нерастворимы в воде, разбавленных растворах щелочей и кислот, не проявляющих свойства окислителей. Растворение ТсО2 в кислых растворах, содержащих Н2О2 или соли CeIV, сопровождается окислением до пертехнетата.
ДОПОЛНЕНИЕ. Ренистый ангидрид
Среди металлов седьмой группы только рений образует стабильный триоксид ReO3, называемый ренистым ангидридом (Сноска: Существование оксида ТсО3 окончательно не доказано.). Это твердое красное вещество с металлическим блеском. В структуре ReO3 атомы рения расположены в центре октаэдров ReO6, каждая из вершин которых сочленена с соседними октаэдрами, образуя бесконечный трехмерный каркас (рис.5.17. Кристаллическая структура ReO3, Уэллс, т.1, стр.253).
Ренистый ангидрид обладает очень низким электрическим сопротивлением, которое, как у типичного металла, уменьшается при понижении температуры. Это объясняется наличием на атоме рения единственного валентного электрона, который делокализован по всей зоне проводимости кристалла.
Оксид рения(VI) синтезируют восстановлением рениевого ангидрида Re2O7 оксидом углерода, металлическим рением или диоксидом:
3 Re2O7 Re = 7ReO3
Ренистый ангидрид относится к кислотным оксидам, однако, обладает низкой химической активностью: он нерастворим в воде и разбавленных растворах щелочей. Образование ренатов происходит лишь при взаимодействии его с горячей концентрированной щелочью:
ReO3 2NaOH = Na2ReO4 H2O.
Длительное кипячение раствора приводит к диспропорционированию рената на перренат NaReO4 и диоксид ReO2. В расплаве щелочи или соды образуются перренат и ренит:
3ReO3 2Na2CO3 = 2NaReO4 Na2ReO3 2СО2↑,
Водными растворами окислителей (HNO3, H2O2, бромная вода) ReO3 переводится в рениевую кислоту:
2ReO3 Н2О2 = 2НReO4.
Для рения известен также голубой пентоксид Re2O5, диспропорционирующий на Re(VII) и Re(IV), но его свойства практически не изучены.
КОНЕЦ ДОПОЛНЕНИЯ
Все три металла образуют высшие оксиды М2О7, однако, по свойствам соединение марганца существенно отличается от оксидов технеция и рения. Марганцевый ангидрид Mn2O7 является одним из наиболее сильных окислителей, в то время как для оксидов Тс2О7 и Re2O7, образующихся непосредственно при горении металлов на воздухе, окислительные свойства практически не характерны.
Марганцевый ангидрид Mn2O7 представляет собой густую летучую гигроскопичную маслянистую жидкость темно-красного цвета, в отраженном свете кажущуюся зеленой с металлическим блеском. При 5,9°C она превращается в красные кристаллы. В безводных серной и фосфорной кислотах оксид марганца(VII) образует оливково-зеленые растворы:
Mn2O7 3H2SO4 2MnO3 3HSO4– H3O
Вещество очень неустойчиво – оно разлагается со взрывом при сотрясении, в присутствии даже следовых количеств органических веществ, а также при нагревании выше 55 °C. Горазно более безопасны в обращении его растворы в четыреххлористом углероде (M. Tromel, M. Russ, Angew. Chem., Int. Ed. Engl., 1987, 26, 1007). Оксид марганца(VII), являющийся ангидридом марганцевой кислоты, получают действием концентрированной серной кислоты на перманганаты щелочных металлов:
2KMnO4 2H2SO4 = 2KHSO4 Mn2O7↓ H2O.
Марганцевый ангидрид один из немногих оксидов металлов, имеющих молекулярное строение. В газообразном, жидком и твердом состоянии он состоит из молекул Mn2O7, представляющих собой два тетраэдра [MnO4], соединенных общей вершиной. В кристаллическом веществе угол Mn-O-Mn составляет 120,7° (A. Simon, R. Dronskowski, B. Krebs, B. Hettich, Angew.Chem., Int. Ed. Engl., 1987, 26, 139) (Рис.5.18. Строение высших оксидов (а) марганца, (б) технеция, (в) рения). Кристаллы, необходимые для определение кристаллической структуры этого вещества, были выращены в тонком стеклянном капилляре, содержащем несколько капель марганцевого ангидрида и сразу помещенном в дифрактометр.
При растворении в воде оксид марганца(VII) Mn2O7 образует темно-фиолетовый раствор марганцевой кислоты НМnO4. Реакция протекает настолько быстро, что над сосудом часто возникает красно-фиолетовый туман, состоящий из капелек раствора НМnO4 и постепенно превращающийся в коричневые частицы пиролюзита .
Марганцевый ангидрид термодинамически нестабилен: он постепенно теряет кислород даже при 0°C. Медленное разложение приводит к образованию γ-MnO2, а при детонации кристаллизуется α-Mn2O3:
Mn2O7 = Mn2O3 2O2↑ ΔH = — 231,3 кДж/моль,
Mn2O7 = 2MnO2 3/2O2↑ ΔH = — 307,1 кДж/моль.
Марганцевый ангидрид окисляет большинство кислородсодержащих органических соединений, во многих случаях происходит воспламенение. На этом основан один из способов зажигания спиртовки в отсутствие спичек. Для этого в фарфоровой чашке растирают несколько кристалликов перманганата калия и приливают к ним две-три капли концентрированной серной кислоты. В полученную маслянистую жидкость, содержащую марганцевый ангидрид, опускают стеклянную палочку, которой затем прикасаются к фитилю спиртовки.
Высшие оксиды технеция и рения представляют собой желтые (Сноска: красноватый оттенок кристаллов Тс2О7 свидетельствует о наличии примеси НТсО4) кристаллические порошки, используемые для очистки и промышленного производства металлов. При нагревании они плавятся без разложения, 120 °C (Тс2О7) и 300 оС (Re2O7), частично переходя в газовую фазу, поскольку обладают высокой летучестью. Высшие оксиды технеция и рения получают окислением металлов, низших оксидов и сульфидов кислородом:
2MS2 7,5O2 = M2O7 4SO2↑,
термическим разложением пертехнетата и перрената аммония:
2NH4MO4 = M2O7 2NH3 H2O.
В газовой фазе эти оксиды стабильны и мономерны, их молекулы состоят из двух сочлененных вершинами тетраэдров МО4. В твердом состоянии такая структура с линейными мостиками сохраняется только у Тс2О7, однако, в отличие от марганцевого ангидрида, угол Тс-О-Тс равен 180° (рис.5.18 б) (B. Krebs, Z. Anorg. Allg. Chem., 1971, 380, 146). Кристаллы высшего оксида рения имеют полимерное строение и состоят из двойных слоев, образованных сильно искаженными октаэдрами ReO6 и правильными тетераэдрами ReO4, соединенными общими вершинами в тетрамерные циклы (рис.5.18 в). Между отдельными слоями существует лишь слабое ориентационное взаимодействие (B. Krebs, A. Muller, H. Beyer, Inorg. Chem., 1969, 8, 436). В каждом октаэдре ReO6 три связи Re-O заметно длиннее трех других и легко разрушаются при нагревании. Этим и объясняется легкая летучесть рениевого ангидрида.
Высшие оксиды технеция и рения, подобно марганцевому ангидриду, чрезвычайно гигроскопичны во влажном воздухе и быстро поглощают воду, превращаясь в технециевую НТсО4 и рениевую НReO4 кислоты, например:
Тс2О7(тв) Н2О(ж) = 2НТсО4(тв) ΔН° = –1,800 кДж/моль, ΔG° = –6,828 кДж/моль.
В отличие от Mn2О7, они являются слабыми окислителями, при комнатной температуре, не способными окислить большинство органических веществ.
Они взаимодействуют со спиртами, пиридином, тетрагидрофураном с образованием комплексных сольватов М2О7.2L (L-молекула орагнического вещества). Даже взаимодействие с амидом калия в жиком аммиаке не сопровождается окислительно-восстановительным процессом, а приводит к образованию нитридорената(VII):
Re2O7 3KNH2 = K2ReO3N KReO4 2NH3.
Оксид Тс2О7 является более сильным окислителем, чем Re2O7. Он восстанавливается парами органических веществ. Таким образом, окислительная активность высших оксидов убывает вниз по группе.
Водородом оба оксида восстанавливаются до металлов. При взаимодействии с монооксидом углерода при 300 атм. и 220 оС технеций образует летучий карбонил Тс2(СО)10, а рений – низшие оксиды ReOх (х < 2), свойства которых практически не изучены.
§
Разнообразие степеней окисления, особенно характерное для марганца, а также амфтерный характер соединений в степенях окисления ниже 5, обусловливает существование большого количества анионных и катионных форм, многие из которых устойчивы в водных растворах. Химия марганца в высших степенях окисления ( 5, 6, 7), наоборот, фактически ограничена манганат-ионами MnO4n–, n = 1 – 3, входящими в состав различных солей. Их традиционные названия, приведенные в таблице 5.3, в соответствии с нормами современной номенклатуры все чаще заменяют термином манганат с указанием степени окисления марганца в скобках.
Таблица 5.3. Типы кислородных соединений марганца в различных степенях окисления.
| Степень окисления | Оксид | Гидроксид | Характер | Катионные формы | Анионные формы |
| 2 | MnO | Mn(OH)2 | основный, слабо амфотерный | MnSO4 | Li2MnO2 Li4[Mn(OH)6] |
| 3 | Mn2O3 | MnOOH Mn2O3×xH2O | амфотерный | Mn2(SO4)3 | LiMnO2 Li3[Mn(OH)6] |
| 4 | MnO2 | MnO2×xH2O | амфотерный | Mn(SO4)2 | Li2MnO3 (манганиты) |
| 5 | – | [H3MnO4] | кислотный | – | Li3MnO4 (гипоманганаты) |
| 6 | – | [H2MnO4] | кислотный | – | Li2MnO4 (манганаты) |
| 7 | Mn2O7 | HMnO4 | кислотный | – | LiMnO4 (перманганаты) |
Степень окисления 2 (d5).
Эта степень окисления наиболее широко представлена у марганца: соединения марганца(II) часто наиболее устойчивы в водных растворах. Химия Тс(II) и Re(II) преимущественно представлена комплексными соединениями, главным образом с фосфинами.
Элменты группы марганца в степени окисления 2 имеют электронную конфигурацию d5, что соответствует наполовину заполненному d-подуровню. Энергетически наиболее устойчивыми оказываются высокоспиновые комплексы (t2g3eg2), в которых достигается большой выигрыш в энергии кристаллизации кристаллическим полем и не требуется затрат энергии на спаривание электронов.
Ион Mn2 в слабом октаэдрическом поле имеет высокосимметричное расположение элетронов, каждый из которых занимает одну из пяти d-орбиталей. Под действием кванта энергии между t2g и eg орбиталями обычно происходят электронные переходы. Однако, в данном случае любой такой переход приведет к спариванию электронов и к понижению симметрии. Поэтому все электронные d-d переходы из высокоспиновой конфигурации d5 со спариванием электронов запрещены по спину, то есть маловероятны:
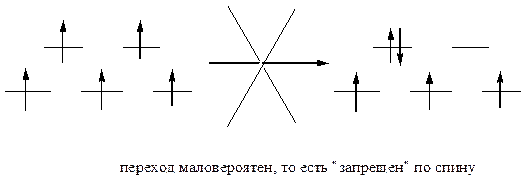
В то же время, как известно, именно d-d переходы и обусловливают окраску ионов переходных металлов. Таким образом, для высокоспиновых октаэдрических комплексов марганца(II) следует ожидать слабую окраску или даже ее отсутствие. В случае искажения октаэдрической геометрии запреты снимаются, и реализуются электронные переходы с появлением интенсивной окраски. Это наблюдается в комплексах с разными лигандами.
Наличие пяти неспаренных электронов вызывает появление парамагнитных свойств ионов. Большинство измеренных магнитных моментов соответствует чисто спиновому значению 5,92 μВ. Такая высокоспиновая октаэдрическая координация реализуется в подавляющем большинстве неорганических и комплексных соединений. Примером может служить акваион [Mn(H2O)6]2 бледно-розового цвета.
В разбавленных водных растворах нитрата марганца(II) гексаакваионы окружены второй более слабо ассоциированной сольватационной оболочкой из 18 молекул Н2О, образующих с внутрисферными молекулами воды систему водородных связей: {[Mn(OH2)6](OH2)18}2 . Гидратная оболочка не зависит от иона нитрата, который не гидратируется. В концентированных растворах вода участвует прежде всего в насыщении первой гидратной оболочки Mn2 , и остающиеся молекулы воды и ионы нитрата статистически размещаются между плотно упакованными комплексами [Mn(H2O)6]2 . В 6,4 молярном растворе Mn(NO3)2 вся вода употребляется для насыщения гидратных оболочек Mn2 , и в этих растворах сохраняется структура твердого Mn(NO3)2.6H2O , содержащая гексакваионы. (Strother E.F., Farach H.A., Poole C.P. Phys. Rev. A, 1971, 4, No 3, 2078).
Высокоспиновые октаэдрические комплексы марганца(II) являются лабильными, поэтому в водных растворах существуют сложные равновесия между аква-ионами, продуктами их гидролиза и замещения молекул воды на анионы кислотных остатков, споосбные выступать в роли лигандов.
В растворах хлорида марганца(II), кроме гидратированного иона Mn2 , присутствуют хлорокомплексы и аквахлорокомплексы общего состава [Mn(H2O)6-nCln]2-n (n = 1, 2, 3), а также продукты гидролиза, прежде всего [Mn(H2O)5OH] (см. ниже.). В сильно разбавленных растворах Mn2 окружен 6 молекулами воды. Начиная с концентрации 0,5 М молекулы воды в гидратной оболочке постепенно замещаются на хлорид-ионы, что приводит к образованию частиц [MnCl(H2O)5] и [Mn(H2O)4Cl2]. При концентрации хлорид-ионов Cl– выше 5M преобладают октаэдрические анионы [MnCl3(H2O)3]—, [MnCl4(H2O)2]2–, [MnCl6]4– а при их огромном избытке – тетраэдрические комплексы [MnCl4]2–. В этих ионах также возможно замещение хлора на молекулы воды: [MnCln(H2O)4-n]2-n (n = 1, 2, 3). (Тишков П.Г., Вишневская Г.П. Журн. Эксперим. и Теор. Физики, 1960, 38, 335).
Гидроксид марганца(II), выпадающий в виде белого осадка при действии щелочей на водные растворы солей марганца(II) (рН осаждения 8,7), на воздухе быстро окисляется:
4Mn(OH)2 O2 = 4MnOOH¯ 2H2O,
поэтому его синтез ведут в инертной атмосфере. Более сильные окислители, такие как бромная вода, гипохлорит, переводят его в пиролюзит:
Mn(OH)2 Br2 2NaOH = MnO2¯ 2NaBr 2H2O
В отличие от большинства гидроксидов переходных металлов, гидроксид имеет стехиметрический состав, отвечающий формуле Mn(OH)2, и изоструктурен гидроксиду магния. Он является основанием средней силы (Kb = 5,0×10–4), превосходящим аммиак, поэтому в присутствии ионов аммония переходит в раствор.
Слабый амфотерный характер гидроксида марганца(II) проявляется в его способности образовывать анионные гидроксо-комплексы:
Mn(OH)2 OH– [Mn(OH)3]–, K ~ 10–5
Так, при взаимодействии с кипящим концентрированным раствором едкого натра в инертной атмосфере образуется тетрагидроксоманганат(II) Na2[Mn(OH)4]. Известны и манганаты(II), полученные твердофазным синтезом, например, K2Mn2O3 (E. Seipp, R. Hoppe, Z. Anorg. Allg. Chem., 1985, 530, 117) (Рис.5.19. Строение K2Mn2O3) , Na14Mn2O9 (G. Brachtel, R. Hoppe, Z. Anorg. Allg. Chem., 1987, 438, 97).
По некоторым свойствам соединения марганца(II) напоминают аналогичные соединения магния, что обусловлено достаточно близкими ионными радиусами (Mn2 0,091 нм, Mg2 0,074 нм), повышенной устойчивостью наполовину заполненного d-подуровня, нулевой энергией стабилизации кристаллическим полем. Сходство между соединениями магния и марганца(II) включает сходные растворимости солей и формулы кристаллогидратов, близкие значения произведений растворимости и констант основности гидроксидов (pKb Mn(OH)2 = 3,30; pKb Mg(OH)2 = 2,60). Подобно магнию, марганец образует белый осадок марганецаммонийфосфата, который при прокаливании переходит в пирофосфат:
MnSO4 (NH4)2HPO4 NH3 H2O = (NH4)MnPO4×H2O¯ (NH4)2SO4
(NH4)MnPO4×H2O = Mn2P2O7 2NH3 3H2O.
Взаимодействием гидроксида марганца(II) с кислотами получают разнообразные соли. В отличие от гидроксида, все они устойчивы к окислению кислородом воздуха. Это легко заметить, сравнивая значения стандарных электродных потенциалов при рН = 0 (E0(Mn3 /Mn2 = 1.51 B) и рН = 14 (E0(Mn(OH)3/Mn(OH)2 = 0.1 B). Соли марганца(II) образуют гидраты, окрашенные в бледно-розовый цвет, свойственный катионам [Mn(H2О)6]2 . Они входят в состав некоторых гидратов, например, MnSO4×7H2O, Mn(ClO4)2×6H2O, Mn(NO3)2×6H2O. В водных растворах солей марганца(II) протекает гидролиз:
[Mn(H2О)6]2 Н2О [Mn(H2O)5OH] H3О , К = 5×10–5
Интересно, что гидролиз растворов галогенидов марганца при нагревании раствора ослабевает, что связано с замещением гидроксид-ионов в координационной сфере металла на галогенид-ионы.
Cульфат марганца(II) в безводном виде представляет собой порошок белого цвета. Его кристаллогидраты образуют розовые кристаллы. Ниже 9 °C из растворов кристаллизуется гептагидрат, содержащий гексаакваионы, между 9 и 24 °C пентагидрат, (марганцовый купорос), изоструктурный медному купоросу, выше 24 °C – моногидрат. Полное обезвоживание наступает лишь при 200 °C. При хранении марганцовый купорос, подобно медному купоросу, выветривается, теряя молекулу внешнесферной воды и, таким образом, превращаясь в тетрагидрат. Безводный сульфат марганца(II) разлагается выше температуры плавления (700 °C):
900 °C
3MnSO4 = Mn3O4 3SO2 O2.
Из сернокислых растворов сульфата марганца(II) кристаллизуется кислая соль Mn(HSO4)2. Известны двойные сульфаты с щелочными металлами, например, K2Mn(SO4)2(H2O)4, K2Mn3(SO4)2(H2O)5.
Действие на растворы солей марганца сульфитами щелочных металлов приводит к осаждению основных солей (см. т.2, стр. 258), например, NaMn2OH(SO3)2H2O (W. Buchmeier, B. Engelen, H.D. Lutz, Z. fuer Naturforsch., 1986, 41, 852). Средний сульфит MnSO3 получают, пропуская ток сернистого газа через водную суспензию карбоната магния.
При осаждении ионов Mn2 растворами сульфидов выпадает осадок телесного цвета, представляющий собой гидратированный сульфид марганца(II) MnS×xH2O. Хотя кристаллическая структура этого продукта неизвестна, бледно-розовая окраска свидетельствует о том, что молекулы воды дополняют координационную сферу марганца до октаэдра. При хранении на воздухе осадок постепенно окисляется, превращаясь в пиролюзит, а в инертной атмосфере теряет кристаллизационную воду, приобретая зеленый цвет, свойственный природному минералу – алабандину (α-MnS), имеющему структуру NaCl. Сульфид марганца (ПР 10–14) растворим даже в разбавленных кислотах и поэтому не может быть осажден сероводородом. Известны также две кристаллические красные модификации, одна – со структурой вюрцита (γ-MnS), другая – сфалерита (β-MnS). β-Модификацию получают, пропуская сероводород в холодный крепкий раствор ацетата марганца(II). При нагревании обе красные модификации переходят в α-MnS. Красно-коричневый дисульфид марганца, или марганцовый колчедан MnS2, изоструктурен пириту. Он образован ионами Mn2 и S22-. Получение дисульфида из простых веществ невозможно, для его синтеза водный раствор сульфата марганца(II) и спиртовой раствор полисульфида натрия помещают в автоклав при 170 оС на несколько суток. Начиная с 250 оС, твердый дисульфид разлагается на α-MnS и пары серы.
Нитрат марганца выделяется из растворов в виде розоватых игольчатых кристаллов гексагидрата Mn(NO3)2×6H2O, растворимых в воде и спирте. При нагревании до 40 °C они превращаются в моногидрат (Рис.5.20. Фазовая диаграмма системы Mn(NO3)2 – H2O), который при 160 °C разлагается:
Mn(NO3)2×H2O ¾® MnO2 2NO2 H2O.
Безводную соль получают обезвоживанием моногидрата азотным ангидридом, выдерживанием его в глубоком ваккууме ниже температуры разложения или взаимодействием марганца с N2O4.
Действием на раствор соли марганца дигидрофосфатом натрия в среде, близкой к нейтральной, удается осадить белый осадок среднего ортофосфата Mn3(PO4)2×7H2O. В сильно щелочной среде осаждаются основные соли, например, Mn2(OH)PO4, Mn5(OH)4(PO4)2. Известны также различные кислые фосфаты, например, Mn(H2PO4)2×H2O, хорошо растворимый в воде (M.D. Marcos, P. Amoros, F. Sapina, D. Beltran, Journ. Alloys Compd., 1992, 188, 133), и малорастворимый MnHPO4×3H2O (Y. Cudennec, A. Riou, Y. Gerault, Acta Cryst. C., 1989, 45, 1411).
При действии на растворы солей марганца(II) раствором карбоната натрия образуется белый осадок карбоната MnCO3, содержащий лишь незначительное количество основной соли. Чтобы избежать ее образование, осаждение проводят гидрокарбонатом. Интересным методом синтеза MnCO3×H2O является взаимодействие растворимой соли марганца с мочевиной в водном растворе при 80 °C. Образующийся в растворе при комнатной температуре комплекс с мочевиной (urea) при нагревании разлагается (E.M. Nour, S.M. Teleb, N.A. Al-Khsosy, M.S: Refat, Transition Metal Chem., 1997, 22, 505):
[Mn(urea)4](NO3)2 6H2O ¾¾® MnCO3×H2O¯ 3CO2 2NH4NO3 6NH3.
Карбонат марганца(II) встречается в природе в виде минерала марганцового шпата. При слабом нагревании (150 °C) в инертной атмосфере он разлагается на оксид MnO и углекислый газ. Если разложение проводить на воздухе, в продуктахм реакции появляется Mn3O4. Растворимость MnCO3 возрастает в 250 раз при насыщении воды углекислым газом. Как и в случае карбонатов магния и щелочноземельных металлов, это объясняется образованием кислой соли, известной лишь в растворе:
MnCO3 CO2 H2O = Mn(HCO3)2.
Силикаты марганца(II) MnSiO3 (родонит), Mn2SiO4 (тефроит) и Mg5(SiO4)2(OH)2 (аллеганиит) встречаются в природе. Они входят в состав шлаков, образующихся в доменном процессе при использовании ферромарганца в качестве раскислителя.
Тетрагидрат ацетатата марганца(II), Mn(CH3COO)2×4H2O, выделяется в виде розово-красных игл из раствора, полученного взаимодействием карбоната марганца с уксусной кислотой. Вещество образовано цепями атомов марганца, соединенных ацетатными мостиками и молекулами воды (Рис.5.21. Строение ацетата марганца(II)).
При взаимодействии суспензии карбоната марганца(II) с горячим раствором щавелевой кислоты осаждается белый кристаллический порошок оксалата марганца MnC2O4×2H2O (известен и тригидрат). При нагревании до 100 °C он теряет воду, а затем разлагается на MnO, CO2 и CO. Растворимость оксалата марганца в воде повышается в присутствии оксалатов щелочных металлов за счет образования анионных комплексов:
Mn2 C2O42– MnC2O4, K = 3×104
MnC2O4, K = 3×104
MnC2O4 C2O42– [Mn(C2O4)2]2–, K = 1×102
[Mn(C2O4)2]2– C2O42– [Mn(C2O4)3]4–, K = 32
В твердом виде получены вещества, содержащие ионы [Mn(C2O4)2(H2O)2]2– и [Mn2(C2O4)3(H2O)2]2–. Интересно, что твердые трисоксалатоманганаты(II) неизвестны.
Устойчивость высокоспиновых комплексных соединений марганца(II), имеющих нулевую энергию стабилизации кристаллическим полем, не всегда коррелирует с положением лиганда в спектрохимическом ряду. Так, например, аммино-комплексы, согласно значениям констант устойчивости, оказываются менее стабильными, чем гидроксо-комплексы. В то же время, для соединений маргнаца(III) стабилизация кристаллическим полем играет существенную роль. Это приводит к ярко выраженной зависимости между окислительно-восстановительной активностью комплексов марагнца(II) и силой лиганда: чем в более сильном поле расположен лиганд, тем менее устойчивым к окислению оказывается комплекс. Если комплексы с лигандами слабого поля, например, [Mn(H2O)4Cl2], K2[Mn(C2O4)2(H2O)2] устойчивы к окислению кислородом воздуха, то аммиакаты и ацетилацетонат марганца(II) на влажном воздухе быстро окисляются. Наблюдается общая закономерность: чем в более сильном поле расположен лиганд, тем менее устойчив к окислению его комплекс с марганцем(II).
При действии на соли марганца(II) избытком водного раствора аммиака выпадает осадок гидроксида, однако часть марганца остается в растворе в виде аммино-комплексов:
[Mn(H2O)6]2 NH3 [Mn(H2O)5(NH3)]2 H2O, K = 0.16.
Ион гексааминомарганца(II) удается зафиксировать лишь в растворе с концентрацией NH3 выше 2М, поэтому этот метод не находит применения в синтезе. Аммиакаты марганца(II) обычно получают в среде жидкого аммиака или пропусканием газообразного аммиака над нагретой твердой солью:
MnCl2 6NH3 = [Mn(NH3)6]Cl2.
Они представляют собой бесцветные порошки, буреющие на воздухе вследствие окисления. В водных растворах часть молекул аммиака замещается на молекулы воды. При взаимодействии марганца с раствором калия в жидком аммиаке образуются амидные комплексы:
Mn 2K 4NH3 = K2[Mn(NH2)4] 2H2↑.
Они довольно устойчивы в сухом виде, но мгновенно разлагаются водой.
Бледно-желтый осадок дигидрата ацетилацетоната марганца(II) Mn(acac)2(H2O)2, полученный в водно-спиртовом растворе по обменной реакции между солью марагнца и ацетилацетонатом щелочного металла, в сухом виде сохраняется долго, однако в присутствии влаги быстро окисляется. При нагревании в вакууме он теряет воду, а затем сублимируется в форме тримера Mn3(acac)6 (Shibata S., Onuma S., Inoue H. Inorg. Chem., 1985, 24, 1723).
Нулевая энергия стабилизация кристаллическим полем означает также, что геометрия образующегося комплекса определяется только стерическими факторами, то есть размером лиганда. Чем крупнее лиганд, тем более вероятно, что комплекс с его участием представляет собой тетраэдр, например, [MnCl4]2–. d-d переходы в тетраэдрических комплексах с конфигурацией d5 более вероятны, чем в октаэдрических, так как тетраэдр не имеет центра симметрии, поэтому для них характерна более интенсивная окраска, обычно желтая или зеленая. Таким образом, отличить октаэдрический комплекс от тетраэдрического часто удается по внешним признакам.
Все рассмотренные выше комплексные соединения являются высокоспиновыми. Спаривание спинов в Mn(II) требует затраты энергии, и поэтому становится благоприятным только под действием лигандов сильного поля, таких как цианид CN– или нитрил CNR. В низкоспиновых комплексах [Mn(CN)6]4- и [Mn(CNR)6]2 с электронной конфигурацией t2g5eg0 из-за сильного π-связывания связь металл-лиганд приобретает существенный ковалентный характер, а центральный атом становится чувствительным к окислению. Темно-фиолетовые кристаллы гексацианоманганата марганца(II) К4[Mn(CN)6] на влажном воздухе превращаются в темно-красный К3[Mn(CN)6]:
4K4[Mn(CN)6] O2 2H2O = 4K3[Mn(CN)6] 4KOH.
Они еще менее устойчивы к окислению, чем гидроксо- и амминокомплексы
Разнообразие комплексных соединений марганца(II) обусловлено высокой устойчивостью высокоспинового состояния с пятью неспаренными электронами. В то же время, у других металлов седьмой группы, расположенных в пятом и шестом периодах, высокоспиновые комплексы не могут существовать из-за высокой энергии расщепления. Как и в случае марганца, стабилизация низкоспинового состояния может быть достигнута преимущественно в комплексах с π-акцепторными лигандами – цианидом, пиридином, нитрилами, фосфинами и арсинами, например, МCl2(py)4, [MCl2(diars)2], где diars — 1,2-диметиларсинобензол, выступающий в роли бидентного хелатирующего лиганда. Часто комплексы технеция(II) и рения(II) сходны между собой по строению и свойствам. В них низшая степень окисления стабилизирована π-связыванием между атомом металла и лигандом.
Большинство таких соединений получают восстановлением комплексов технеция(III) и рения(III), например, фосфорноватистой кислотой Н3РО2 , хлоридом олова(II), электрохимически.
При восстановлении гексахлоротехнетата(IV) гидроксиламином образуется комплексный катион Тс(II):
[TcCl6]2- 4NH2OH 5NH3 = [Tc(NH2OH)2(NH3)3]2 6Cl— N2↑ 2H2O 2NH4 .
Многие комплексные соединения технеция(II) и рения(II) стабилизированы за счет образования кратных связей металл-металл. Так, при обработке иона [Tc2Cl8]2- тетрафтороборной кислотой HBF4 в ацетонитриле выделен синий комплекс [Tc2(CH3CN)10](BF4)4 с тройной связью Тс≡Тс длиной 2,122 А. Фосфиновые лиганды способны стабилизировать низкие степени окисления путем делокализации электронной плотности от металла на лиганды. Благодаря высокой электронной плотности между атомами металла эти комплексы участвуют в восстановлении органических соединений, в реакциях окислительного присоединения. В твердом виде они медленно реагируют с кислородом, окисляясь вплоть до степени окисления 5 без изменений в координационной сфере. (J. Inorg. Chem., 1995, 34, 1880 ) Авторы ???
Рис.5.22. Строение комплексов [Tc2(CH3CN)10]4–, Re2Cl4(μ-Ph2PCH2PPH2)
Степень окисления 3d4).
Марганец в степени окисления 3 может быть стабилизирован в виде оксида и гидроксида, практически нерастворимых в воде, а также в форме разнообразных комплексных соединений, устойчивость которых возрастает по мере увеличения силы лиганда. Большинство этих комплексов октаэдрические и высокоспиновые, независимо от входящих в их состав лигандов. Поэтому магнитные моменты комплексов близки к чисто спиновому значению 4,90 μВ для четырех неспаренных электронов.
Электронная конфигурация d4 иона Mn3 , относящегося к ян-теллеровским ионам, приводит к искажению октаэдрических комплексов, преимущественно за счет удлинения двух связей, расположенных в транс-положении друг к другу. Все комплексы марганца(III) лабильные.
Гексаакваион [Mn(H2O)6]3 образуется при анодном окислении солей марганца(II), а также при восстановлении водных растворов перманганатов. По-видимому, он является интермедиатом во многих окислительно-восстановительных превращениях, включая окисление Mn2 персульфатом. В твердом виде он охарактеризован в цезий-марганцевых квасцах [Cs(H2O)6][Mn(H2O)6](SO4)2, кораллово-красный цвет которых связывают с окраской этого иона. (Сноска: в структуре квасцов ян-теллеровское искажение иона [Mn(H2O)6]3 не обнаружено). Подобно другим трехзарядным катионам металлов первого переходного ряда, в водных растворах он значительно гидролизован:
[Mn(H2O)6] 3 H2O [Mn(H2O)5OH] 2 H3O , K = 0.93.
Возможно, при этом образуются также и полиядерные гидроксокомплексы. При попытке концентрирования растворы мутнеют вследствие образования аморфного гидроксида переменного состава. Высказано предположение, что грязно-зеленый цвет растворов, образующихся при анодном окислении солей марганца, как раз и обусловлен окраской не гексаакваиона, а продуктов его гидролиза.
Даже разбавленные водные растворы, содержащие [Mn(H2O)6]3 ( E0(Mn3 /Mn2 = 1.56 B) неустойчивы вследствие окисления им воды:
4[Mn(H2O)6] 3 2H2O = 4[Mn(H2O)6]2 4H O2.
Параллельно с этим, как видно из диаграммы Фроста (Рис.5.2), возможно также его диспропорционирование:
2[Mn(H2O)6] 3 = [Mn(H2O)6] 2 MnO2¯ 4H 4H2O.
Гидроксид марганца(III) образуется в виде бурого осадка переменного состава при окислении гидроксида марганца(II) кислородом воздуха. Первоначально образующийся продукт содержит атомы марганца в двух степенях окисления — 2 и 3 и отвечает составу MnOx×nH2O, где x = 1.35 – 1.44, примерно соответствующему гидратированному гауcманниту (В.П. Чалый, Гидроокиси металлов, Киев. Наукова думка, 1972, с. 34). При стоянии он превращается в форму оксогидроксида MnOOH, известного в виде двух модификаций – ромбической (γ-MnOOH, манганит) и моноклинной (α-MnOOH, гроутит).
В структуре манганита каждый атом марганца находится в центре октаэдра MnO6. Отдельные октаэдры объединены друг с другом в гофрированные слои, соединенные водородными связями, в которых участвуют только ОН-группы: -O-H…O(H)-. Такое же строение имеют бемит γ-AlOOH и лепидокрокит γ-FeOOH. В структуре гроутита октаэдры MnO6 соединены общими ребрами в двойные цепи, сочлененные отдельными вершинами в трехмерный каркас и дополнительно сшитые водородными связями. В отличие от структуры γ-модификации, здесь они образованы между гидроксилом и оксо-группой: -O…H-O. Аналогичное строение имеют диаспор α-AlOOH, монтрозеит α-VOOH, гетит α-FeOOH.
Рис.5.23. Строение (а) α-MnOOH, (б) γ-MnOOH
§
Препаративный метод синтеза манганита состоит в окислении сульфата марганца 3 %-ным раствором пероксида водорода в аммиачной среде:
2MnSO4 H2O2 4NH3 2H2O = 2MnOOH¯ 2(NH4)2SO4
В этой реакции окислителем выступает также кислород, образующийся при каталитическом разложении пероксида на частицах продукта. Осадок, отфильтрованный и высушенный при 100 °C, имеет состав MnOOH.
Оксогидроксид марганца(III) является амфотерным основанием. При кипячении его со щелочами образуются красные растворы, из которых кристаллизуются гидроксоманганаты(III), например, Na3[Mn(OH)6].2,5H2O и Na4[Mn(OH)7].5H2O, Ba2[Mn(OH)7] и Ba[Mn(OH)5]. В твердой фазе получены соединения состава NaMnO2, Na4Mn2O5, Ca2Mn2O5, Ca4Mn2O7 (K.R. Poepplmeier, M.E. Leonowicz, J.M. Longo, J. Solid State Chem., 1982, 44, 89).
При растворении MnOOH на холоду в крепкой серной кислоте образуется красный раствор, из которого могут быть выделены красные кристаллы кислого сульфата Mn2(SO4)3×H2SO4×8H2O, на самом деле представляющие собой анионный комплекс H5O2 [Mn(SO4)2(H2O)2]– (F.M. Cgang, M. Jansen, D. Schmitz, Acta Cryst. C, 1983, 39, 1497). Перекристаллизация из концентрированной серной кислоты, действующей в качестве водоотнимающего средства, приводит к гигроскопичным темно-зеленым игольчатым кристаллам среднего сульфата Mn2(SO4)3 неизвестного строения. Он представляет собой ковалентное соединение, растворимое в серной кислоте, но гидролизующееся при разбавлении этого раствора водой (J.B. Vincent, H.R. Chang, K. Falting, J.C. Huffman, G. Christou, D.N. Hendrickson, J. Amer. Chem. Soc., 1987, 109, 5703). При контролируемом гидролизе может быть получен основный сульфат Mn(OH)SO4(H2O)2 (K. Mereiter, Acta Cryst. B., 1979, 35, 579).
Наиболее распространенным методом получения солей марганца(III) служат окислительно-восстановительные превращения. Так, сульфат марганца(III) может быть получен также анодным окислением горячего раствора сульфата марганца(II) в серной кислоте:
эл. ток
2MnSO4 H2SO4 ¾¾¾® Mn2(SO4)3 H2
или восстановлением перманаганата калия серной кислотой:
2KMnO4 4H2SO4 = Mn2(SO4)3 K2SO4 2O2 4H2O.
Если тонкий порошок KMnO4 обработать ледяной уксусной кислотой, постепенно происходит его восстановление, ускоряющееся при нагревании. Около температуры кипения интенсивно выделяется СО2. Если образовавшийся красно-коричневый раствор смешать с концентрированной серной кислотой до растворения осадка и прокипятить, то при охлаждении из него выпадают красные кристаллы квасцов,
2KMnO4 CH3COOH 4H2SO4 6H2O = 2KMn(SO4)2×12H2O ↓ 2CO2↑,
В присутствии воды они нацело гидролизуются, превращаясь в оксогидроксид марганца(III), а при температуре выше 0 °C разрушаются. Устойчивость квасцов возрастает с увеличением размера катиона щелочного металла, но даже цезиевые квасцы разлагаются при температуре чуть выше комнатной.
Нитрат марганца(III) в чистом виде не описан, однако известны его аддукты с азотдонорными лигандами, например, Mn(NO3)3phen и анионный нитрониевый комплекс NO2 [Mn(NO3)4]–. Синтезировано большое количество фосфатов марганца(III), например, серо-зеленый MnPO4×H2O, красно-фиолетовый Mn(PO3)3 (M. Bagieu-Beucher, Acta Cryst. B., 1978, 34, 1443), красный, растворимый в воде NaMn(HPO4)2 и разнообразные фиолетовые пирофосфаты, образующиеся в окислительном пламени при получении фосфатных перлов с солями марганца(II):
2NaNH4HPO4 Mn(NO3)2 = NaMnP2O7 2NH3 NaNO3 NO2 2H2O.
Ранее они использовались в качестве пигментов. Доказано, что при действии окислителей (KMnO4, HNO3) на раствор соли марганца(II) в присутствии избытка пирофосфат-ионов образуются фиолетовые растворы, в которых в умеренно кислой среде доминирует комплекс [Mn(H2P2O7)3]3–.
Оксалат марганца(III) существует только в форме комплексных соединений. Исходный комплекс готовят in situ , восстанавливая перманаганат избытком щавелевой кислоты и добавляя поташ как дополнительный источник ионов K :
2KMnO4 11H2C2O4 3K2CO3 = 2K4[Mn(C2O4)3] 13CO2 11H2O,
4K4[Mn(C2O4)3] KMnO4 4H2C2O4 = 5K3[Mn(C2O4)3] 4H2O K2C2O4
Другим методом синтеза является растворение оксогидроксида марганца(III) в растворе, cодержащем щавелевую кислоту и оксалат калия (Bhattacharjee M.N., Chaudhuri M.K., Dutta Purkayastha R.N. Inorg. Chem., 1985, 24, 447):
2MnOOH 3H2C2O4 3K2C2O4 = 2K3[Mn(C2O4)3] 4H2O.
Образующиеся при охлаждении красно-фиолетовые кристаллы настолько чувствительны к свету, что все работы с ними, включая синтез, следует проводить в темноте. На свету происходит внтуримолекулярная окислительно-восстановительная реакция:
2K3[Mn(C2O4)3] = 2MnC2O4 3K2C2O4 2CO2
Водные растворы комплекса даже в отсутствие света становятся желтыми вследствие реакции замещения:
K3[Mn(C2O4)3] 2H2O K[Mn(C2O4)2(H2O)2] K2C2O4
Ион [Mn(C2O4)2(H2O)2]– существует в виде двух изомеров – зеленого (цис-) и желтого (транс-) (W. Levason, C.A. McAuliffe, Coord. Chem. Rev., 1972, 7, 353).
Ацетилацетонат марганца(III) Mn(acac)3 получают взаимодействием ацетилацетона с крепким водным раствором перманганата калия. Комплекс, выделяющийся в виде коричневого осадка, очищают сублимацией. Он состоит из октаэдрических молекул.
Гексацианоманганат(III) калия получают окислением гексацианоманганата(II) перманаганатом калия. Темно-красный гексацианоманганат(III) K3[Mn(CN)6], образующийся при продувании воздуха через водный раствор соли марганца(II), в который добавлен избыток цианистого калия, или при растворении карбоната марганца в водном растворе цианида под действием пероксида водорода
2MnCO3 12KCN H2O2 = 2K3[Mn(CN)6] 2K2CO3 2KOH,
является редким примером низкоспинового комплекса марганца(III). Интересно, что в отличие от большинства других гексацианометаллатов, он лабилен и поэтому легко разлагается водой до MnOOH. При действии на гексацианокомплекс раствором хлорной кислоты выделяется осадок Mn(CN)3, который, как показано (B. Ziegler, M. Witzel, M. Schwarten, D. Babel, Z. Naturforschung, Teil. B., 1999, 54, 870), представляет собой гексацианоманаганат(IV) марганца(II) MnII[MnIV(CN)6] (Рис.5.24. Строение MnII[MnIV(CN)6]). Это является еще одним доказательством неустойчивости соединений марганца(III) к диспропорционированию.
Для марганца(III) известны также и многоядерные комплексы, в которых роль мостиковых лигандов выполняют атомы кислорода. К простейшим примерам относится желто-коричневый диядерный цианокомплекс K7[Mn2O(CN)10], образующийся в качестве побочного продукта при получении гексацианоманаганата(III).
Основный ацетат марганца(III), долгое время считавшийся “дигидратом Mn(CH3COO)3×2H2O”, на самом деле представляет собой трехъядерный оксокомплекс [Mn3O(CH3COO)6(H2O)6] CH3COO– (C.L. Raston, A.H. White, A.C. Willis, K.S. Murray, Journ. Chem. Soc., Dalton Trans., 1974, 1793), построенный аналогично другим оксоацетатам трехвалентных металлов (алюминия, см. том 2, с. 100, ванадия, см. том 3, с., хрома, см. том 3, стр.) Коричневые, с шелковистым блеском кристаллы этой соли получают окислением ацетата марганца(II) в ледяной уксусной кислоте перманганатом:
3KMnO4 12Mn(CH3COO)2 14CH3COOH 23H2O = 5Mn3O(CH3COO)7(H2O)6 3CH3COOK.
Безводная соль, образующаяся в виде коричневого порошка при взаимодействии нитрата марганца(II) с уксусным ангидридом
6Mn(NO3)2 8(CH3CO)2O H2O = 2Mn3O(CH3COO)7 12NO2 3/2O2 2CH3COOH,
по-видимому, также содержит в своем составе трехъядерные фрагменты [Mn3O(CH3COO)6(H2O)6] , связанные друг с другом ацеататными мостиками. Оба вещества устойчивы при хранении в отсутствии влаги, но в воде необратимо гидролизуются. Их часто используют в лабораторной практике для синтеза других соединений марганца(III) по реакциям обмена.
Еще более сложное строение имеют карбоксилатные комплексы марганца со смешанными степенями окисления. Соединение Mn12O12(CH3COO)16(H2O)4 , впервые полученное в виде темно-красных кристаллов в виде примеси к «дигидрату», образующейся при использовании избытка перманганата калия (P.D.W. Boyd, Q. Li, J.B. Vincent et al, Journ. Amer. Chem. Soc., 1988, 110, 8537; V. McKee, Adv. Inorg. Chem., 1994, 40, 323; G. Christou et al, Inorg. Chem., 1991, 30, 1665; G. Christou et al, Journ. Amer. Chem. Soc., 1993, 115, 1804), можно представить как центральный куб Mn4IVO4, построенный из четырех атомов марганца(IV) и четырех атомов кислорода, к которому присоединены 16 μ2-мостиковых ацетатных групп и 12 μ3-мостиковых атомов кислорода. Из двенадцати атомов марганца четыре центральных имеют степень окисления 4, а восемь остальных 3. (Рис.5.25. Строение Mn12O12(CH3COO)16(H2O)4). Часть ионов Mn3 в структуре комплекса может быть замещена на ионы Fe3 до состава [Mn8Fe4O12(CH3COO)16(H2O)4]. Благодаря обменному взаимодействию между атомами металла, входящими в состав молекулы, их магнитные моменты суммируются и действуют как единый «суперспин». Такого рода соединения представляют собой перспективные молекулярные магнитные материалы (Sessoli R., Gatteschi D., Caneschi A., Novak M.A., Nature, 1993, 365, 141).
Все соединения марганца(III), особенно образованные лигандами слабого поля и хорошо растворимые в воде, являются сильными окислителями. Они выделяют иод из иодида калия, окисляют сероводород до серы:
Mn2(SO4)3 2KI = 2MnSO4 I2 K2SO4
Только сильные окислители в щелочной среде переводят их в манганаты(VI).
Химия технеция и рения в низших степенях окисления сильно отличается от химии соединений марганца наличием большого числа соединений со связью металл-металл и практически полным отсутствием простых катионных форм. Гидратированные оксиды, по составу приближающиеся к дигидратам, получают гидролизом галогенидов в отсутствии кислорода воздуха. Гидратированный оксид рения(III) постепенно разлагает воду с выделением водорода, а при попытке обезвоживания диспропорционирует.
Химия технеция(III) и рения(III) представлена в основном галогенидами и комплексными соединениями. Подобно марганцу, эти металлы образуют октаэдрические комплексы. Обычно их получают восстановлением пертехнетатов и перренатов. Наиболее устойчивы комплексы с π-акцепторными лигандами, например, фосфинами.
Исходным веществом для синтеза разнообразных комплексных соединений рения служит аддукт оксохлорида с трифенилфосфином, образующийся в виде желтых призматических кристаллов при кипячении рениевого ангидрида или перрената калия с соляной кислотой и трифенилфосфином в водно-спиртовом растворе:
2KReO4 8HCl 6PPh3 = 2ReOCl3(PPh3)2 2PPh3O 2KCl 4H2O.
При взаимодействии его с первичными аминами образуются иминокомплексы, с фенилгидразоном – хелат, а с уксусным ангидридом – диядерный ацетат, построенный аналогично ацетату хрома(II) Cr2(CH3COO)4(H2O)2, но содержащий вместо воды координированные атомы хлора:
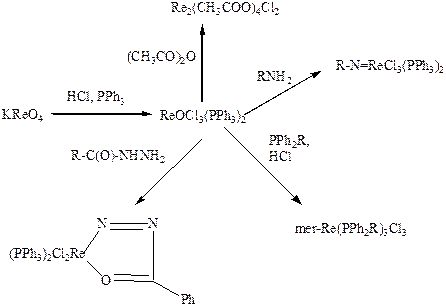
Аналогичное строение имеет диядерный анион, обнаруженный в двойном сульфате рения(III)-аммония (NH4)2Re2(SO4)4(H2O)2 (Рис. 5.26. Строение [Re2(SO4)4(H2O)2]2–) (Козьмин П.А., Ларина Т.Б., Суражская М.Д., Котельникова А.С., Османов Н.С., Коорд. хим., 1980, 6, 1264). Эта соль образуется при растворении димерного трихлоромоноформиаторената(III) аммония в H2SO4. Димерные анионы содержат четверную связь Re-Re (0,2217 нм)и соединены мостиковыми сульфатными группами (Re-O 0,202 нм).
Некоторые комплексы способны сохранять состав и строение координационной сферы при окислительно-восстановительных превращениях. Это используют в синтезе. Например, роданотехнетаты(III) [Tc(NCS)6]3- получают восстановлением [Tc(NCS)6]2- гидразином.
Кроме октаэдрических, известны комплексы с другими координациями центрального атома: например, 7-координированный [M(CN)7]4-
Многие комплексы Тс(III) и Re(III) легко окисляются до высших степеней окисления. Кластеры, подобные описанному выше ацетату рения(III), обладают повышенной устойчивостью, благодаря кратной связи металл-металл.
Степень окисления 4 (d3).
Данная степень окисления для марганца представлена сравнительно небольшим числом соединений, премущественно комплексных. Стабилизации Mn(IV) способствуют сильно электроотрицательные анионы, такие как F–, O2–, полидентатные анионы, содержащие элементы в высших степенях окисления (например, периодаты), либо объемные изополианионы ванадия, ниобия, молибдена, вольфрама, образующие с марганцем гетерополисоединения. Наличие у марганца(IV) трех d-электронов (он изоэлектронен Cr3 ) обусловливает инертность его соединений, что приводит и к их кинетической стабильности. По этой же причине синтез комплексов марганца(IV) лишь в редких случаях удается провести по обменным реакциям, как правило, для этого требуются окислительно-восстановительные превращения. Большинство комплексов октаэдрические и имеют чисто спиновый магнитный момент 3,87 М.Б., что соответствует трем неспаренным электронам.
Наиболее устойчивым соединением марганца(IV), безусловно, является диоксид MnO2, описанный выше. Образуемые им гидраты, например, MnO2×1/2H2O, встречающийся в природе в виде минерала тодорокита, легко теряют воду и по причине химической инертности не вступают в обменные реакции с кислотами и основаниями. О его амфотерности можно судить косвенно по наличию как катионных, так и анионных форм. Ионы гексааквамарганца(IV) [Mn(H2O)6]4 , подобно другим четырехзарядным гексааквакатионам 3d-металлов, в водных растворах неустойчивы вследствие необратимого гидролиза и высокой окислительной активности. Однако об их окраске мы можем судить по другим соединениям, содержащим марганец(IV) в октаэдрическом поле, образованном атомами кислорода. Примерами служат периодат, образующийся при добавлении сульфата марганца(II) к подкисленному раствору тригидропериодата натрия
MnSO4 2Na2H3IO6 = NaMnIO6↓ NaIO3 Na2SO4 3H2O
либо 9-молибдоманганат(IV) аммония (NH4)6MnMo9O32×6H2O, получаемый из гептамолибдата, перманганата калия и пероксида водорода. Оба эти вещества представляют собой оранжево-красные кристаллические порошки.
При взаимодействии свежеосажденного гидратированного диоксида марганца с горячей 75%-ной иодноватой кислотой из образующегося темнокоричневого раствора выпадает красный осадок комплексной кислоты H2MnIV(IO3)6.2H2O. Получены ее соли M2Mn(IO3)6 c щелочными металлами и аммонием. (Мележик А.В., Павлов В.Л. Журн. Неорган. Химии, 1975, 20, 678). Известны и более сложные периодатные комплексы, например, Na7Mn(HIO6)2(H2IO6)×18H2O (W. Lewason, M.D. Spicer, M. Webster, Inorg. Chem., 1992, 31, 2575). Во всех этих соединениях атом марганца расположен в центре правильного октаэдра MnO6, образованного атомами кислорода иодат- или периодат-ионов, выступающих в роли хелатирующих лигандов (Рис.5.27. Строение аниона в Na7Mn(HIO6)2(H2IO6)×18H2O).
Неорганические соли марганца(IV) крайне неустойчивы и мало изучены. Так, сульфат марганца(IV) Mn(SO4)2, выделяется в вид е черных кристаллов из достаточно крепких сернокислых растворов при добавлении в них сульфата марганца(II) и перманганата калия (Руководство по препаративной неорганической химии, под ред. Г. Брауэра, М., Изд-во Иностр. Лит., 1956, с. 671):
3MnSO4 2KMnO4 8H2SO4 = 5Mn(SO4)2 K2SO4 8H2O.
Он растворим в 70%-ной серной кислоте с образованием темного раствора, но при разбавлении водой необратимо гидролизуется:
Mn(SO4)2 2H2O = MnO2¯ 2H2SO4.
Фосфат марганца(IV) известен только в виде комплексов [Mn(PO4)2]2–, существующих при высоких концентрациях ортофосфорной кислоты. Любопытным исключением является малорастворимый в воде селенит марганца(IV) Mn(SeO3)2, оранжево-красных кристаллы которого образуются с высоким выходом при взаимодействии пиролюзита или перманаганата калия с диоксидом селена в кипящей воде! Лишь при 400 °C вещество диспропорционирует на селенат марганца(IV) и селенистый ангидрид.
Амфотерный характер гидратированного оксида марганца(IV) проявляется в способности образовывать оксо- и гидроксоанионы, называемые манганатами(IV) или манганитами.
Так, при сплавлении пиролюзита с оксидом кальция получены соединения CaMnO3, Ca2MnO4, Ca2Mn3O8, Ca4Mn3O10 (K.R. Poepplmeier, M.E. Leonowicz, J.C. Scanlon, J.M. Longo, W.B. Yelon, Journ. Solid State Chem., 1982, 45, 71; 1985, 58, 71). Практически во всех манганитах атом марганца находится в октаэдрическом окружении, исключение составляет манганит бария Ba3MnO5, в структуре которого присутствуют тетраэдры MnO4. При нагревании манганиты диспропорционируют:
: 500°C
2K2MnO3 = K3MnO4 KMnO2.
(Рис. 5.28. Диаграмма системы CaO-MnO2 НАЙТИ!!! МОЖНО ЗАМЕНИТЬ Ca на что-то). Гидроксоманагнатат(IV)-ион трудно получить в растворе, но он известен в природном минерале журавските Ca3[Mn(OH)6](CO3)(SO4).13H2O ярко-желтого цвета.
ДОПОЛНЕНИЕ. Пероксосоединения марганца(IV)
В низших степенях окисления марганец не образует пероксидных соединений, однако пероксоманганаты(IV) известны (Сноска: раньше диоксид марганца ошибочно называли «перекисью марганца» по аналогии с пероксидом бария, имеющим такой же состав. Такое название можно встретить в старой литературe). Они играют важную роль в в процессе фотосинтеза, являясь интермедиатами при окислении воды марагнецсодержащим олигопротеином, а затем распадаясь с выделением кислорода. Марганец присутствует в фотосистеме II, где поглощение излучения обеспечивает энергию для окисления воды, выделения СО2 и переноса электронов в фотосистему I. Здесь осуществляется восстановление никотинамиддинуклеотидфосфата. При этом окисление Mn(II) до Mn(III) и Mn(IV) протекает через образование гидроксо- и пероксокомплексов (Рис.5.29. Предполагаемый механизм окисления воды активным центром марганец-содержащего олигопротеинового комплекса. Показан на модельном соединении с лигандом – 1,4,7-триметил-1,4,7-триазациклононаном). Считается, что «окисляющие воду центры» фотосистемы II содержат по четыре атома Mn в виде плоских или кубановых кластеров, однако точное строение марганец-содержащего олигопротеинового комплекса неизвестно. В последние годы в центре внимания оказались пероксокомплексы марганца со многими модельными соединениями, имитирующими строение активных центров, а также с порфиринами и фталоцианинами (K. Weighardt, Angew. Chem., Int. Ed. Engl., 1989, 28, 1153; V.K. Yachandra, K. Sauer, M.P. Klein, Chem. Rev., 1996, 96, 2927).
Простейшие пероксосоединения марганца изучены гораздо хуже, так как не являлись предметом современных исследований. Доказано (R. Scholder, A. Kolb, Z. Anorg. Allg. Chem., 1949, 260, 231), что при действии манганата(VI) или cоли марганца(II) на охлажденный раствор пероксида водорода в щелочной среде осаждается коричневый кристаллический пероксоманганат(IV) K3[Mn(OH)(O2)3], строение которого неизвестно. Есть сообщения о получении и других пероксокомплексов: K2[Mn(H2O)(O2)3], K4[Mn(O2)4]. Все они неустойчивы и быстро распадаются, выделяя кислород и превращаясь в диоксид марганца. Каталитическое действие соединений марганца на разложение пероксида водорода также, очевидно, связано с образованием неустойчивых пероксосоединений.
КОНЕЦ ДОПОЛНЕНИЯ
ДОПОЛНЕНИЕ. Гетрополисоединения марганца.
Благодаря электронной конфигурации d3, не вызывающей искажения октаэдрического поля, марганец(IV), технеций(IV) и рений(IV) выступают в качестве гетероатомов в большом количестве гетерополисоединений.
Схема образования и взаимных переходов между различными типами гетерополиванадатоманганатов(IV) изображена на рис.5.30. Общий способ их синтеза заключается в обработке нагретых смесей растворов KVO3 и MnSO4 персульфатами щелочных металлов или аммония при различных рН. Выделены красные кристаллы трех типов: тетраванадоманганаты(IV) [K2MnV4O13•4H2O, (NH4)5HMn3V12O39•nH2O], 11-ванадатоманганаты(IV) [K5MnV11O32•nH2O, Cs4,5H0,5Mn11V32•7H2O] и 13-ванадатоманганаты(IV) [K7MnV13O38•18H2O, Na7MnV13O38•29H2O]. Изменения соотношений реагентов и кислотности растворов приводят к взаимным переходам этих соединений друг в друга (рис.5.29). При подщелачивании растворов гетерополианионы разрушаются.
Продуктами взаимодействия ниобатов с ацетатом натрия и пероксидом водорода в щелочной среде являются гетерополи-12-ниобоманганаты(IV). В соединении Na12MnΝb12O38•50H2O анион MnNb12O3812- построен из центрального октаэдра MnO6, окруженного двумя группами Nb6O19, составленными из октаэдров NbO6 (рис.5.31. Структура аниона MnNb12O3812- в Na12MnNb12O38.50H2O). Среднее расстояние Mn-O 0,187 нм, точно такое же, как в периодате NaMnIO6.
При реакциях кипящих растворов парамолибдатов, паравольфраматов или мономерных вольфраматов с MnSO4 и окислителями (персульфатом или пероксидом водорода), образуются несколько типов гетерополисоединений, кислот или их солей. Свободная кислота H6MnMo9O32 известна только в растворе, однако выделены ее кристаллические соли со щелочными металлами, 9-молибдоманганаты(IV), например, [(NH4)6MnMo9O32•nH2O]. При кипячении растворов Na2MoO4 и NaMnO4 с соляной кислотой образуется желтый раствор, из которого введением избытка хлорида аммония осаждают желтые кристаллы 12-молибдоманганата(IV), (NH4)3HМnMo12O40. При повышении рН растворов соединения полностью разрушаются с осаждением диоксида MnO2 и образованием растворимых молибдатов. Аналогично получены 12-вольфрамоманганаты(IV), например, K4MnW12O40•20H2O. Описаны и другие типы гетерополисоединений, в частности, пентавольфрамоманганаты(IV), Na6MnW5O20•18H2O.
КОНЕЦ ДОПОЛНЕНИЯ
Многие комплексные соединения Mn(IV) обладают сильными окислительными свойствами. Это неудивительно, так как для более высоких степеней окисления марганца комплексы нехарактерны. Цианидный комплекс марганца(IV) K2[Mn(CN)6] желтого цвета образуется при окислении гексацианоманганата(III) хлористым нитрозилом в диметилформамиде. Он является сильным окислителем, неустойчивым в водном растворе. Ацетилцетонат марганца(IV) известен только в форме неустойчивого катиона [Mn(acac)3] , образующегося при электрохимическом окислении трис-ацетилацетоната марганца(III) в ацетонитриле.
Для марганца характерно образование многоядерных комплексов, в состав которых одновременно входит несколько атомов металла. Такие соединения обычно стабилизируются оксо- и ацетатными мостиками, а также ди – и полиаминами. Простейший диядерный оксалатный комплекс получают аналогично трис-комплексу марганца(III), но при другом соотношении реагентов (Y. Yoshino, I. Taminaga, M.Kokiso, T. Uehiro, Bull. Chem. Soc. Jаpаn, 1974, 47, 2787):
2KMnO4 6H2C2O4 K2C2O4 = K4[Mn2O2(C2O4)4] 3CO2 6H2O.
Выделяющиеся зеленые кристаллы тетрагидрата устойчивы при пониженной температуре в темноте в течение недели. (Рис.5.32. Строение аниона [Mn2O2(C2O4)4]4–).
Черно-коричневые гидратированные диоксиды технеция TcO2.2H2O и рения ReO2.2H2O осаждаются при восстановлении водных растворов пертехнетатов и перренатов водородом в момент выделения, солями олова(II) или электрическим током. Они не имеют постоянного состава и в водных растворах не проявляют ярко выраженных кислотно-основных свойств. Дегидратацией в вакууме при 250-300 оС их переводят в оксиды. Другим методом синтеза служит гидролиз хлоридных комплексов K2TcCl6, K2ReCl6 :
K2TcCl6 H2O = TcO2.2H2O 2KCl 6HCl,
осуществляемый при кипячении в инертной атмосфере, а для диоксида рения — также и гидролиз высших хлоридов ReCl4 и ReCl5:
3ReCl5 12H2O = 2ReO2.2H2O HReO4 15HCl.
При обработке дигидрата ТсО2.2Н2О концентрированными растворами щелочей образуются оранжевые растворы, содержащие гидроксокомплексы [Тс(ОН)6]2–:
TcO2.2H2O 2KOH = K2[Tc(OH)6].
Онилегко окисляются в пертехнетаты пероксидом водорода, бромной водой, солями церия(IV). Диоксид рения ReO2 устойчив к действию растворов щелочей, но при сплавлении в вакууме реагирует с ними. В продуктах реакции найдены коричневые рениты, например, Li2ReO3 (R.J. Cava, A. Santoro, D.W: Murphy, S. Zahurak, R.S. Roth, J. Solid State Chem., 1982, 42, 251). Если сплавление проводить на воздухе, образуются перренаты. По сравнению с диоксидом марганца, гидратированные оксиды технеция(IV) и рения(IV) гораздо более слабые окислители, о чем свидетельствует их взаимодействие с соляной кислотой, приводящее к хлоридным комплексам Н2МCl6 (см. раздел оксиды). Восстановительные свойства диоксидов технеция и рения выражены значительно сильнее, чем у MnO2: сильные окислители (HNO3, KMnO4, H2O2, HClO3) переводят их в анионы технециевой и рениевой кислот:
§
MnO4– 4H 3e– ® MnO2 2H2O, E0 = 1,70 B
TcO4– 4H 3e– ® TcO2 2H2O, E0 = 0,74 B
ReO4– 4H 3e– ® ReO2 2H2O, E0 = 0,51 B
Для химии технеция(IV) и рения(IV) практически не характерно наличие катионных форм, поэтому простые соли этих металлов, несмотря на устойчивость этой степени окисления как к окислению, так и к восстановлению, неизвестны. Большинство изученных соединений представляют собой комплексы, например, [ReCl6]2–, [Tc(NCS)6]2–, Re2Cl4(acac)4 и K4[Re2O2(C2O4)4], в котором два атома металла соединены между собой двумя оксо-мостиками и связью металл-металл (Рис.5.33. Строение аниона в [Re2O2(C2O4)4]4–) (Lis I., Acta Cryst. B., 1975, 31, 1594). Поэтому здесь расстояние Re–Re (0,236 нм) оказывается короче, чем в металлическом рении (0,274 нм). Комплексные соединения рения, как правило, более устойчивы к гидролизу, окислению, разложению, чем аналогичные соединения технеция.
ДОПОЛНЕНИЕ. Дисульфид рения
Дисульфид рения ReS2 представляет собой черный порошок, устойчивый на воздухе, нерастворимый в воде, растворах щелочей, соляной и серной кислот, сульфидов щелочных металлов. Он начинает формироваться из простых веществ уже при комнатной температуре, но реакция завершается при нагревании до 1000 оС. Из других путей синтеза дисульфида можно использовать восстановление высшего сульфида в токе Н2 уже при комнатной температуре, сплавление перрената КReО4 с серой и содой, нагревание оксидов ReО3 или ReО2 с серой или в атмосфере Н2S. В структуре ReS2 атомы расположены между плотно упакованными слоями из атомов серы (Рис.5.34. Строение ReS2). (Murray H.H., Kelty S.P., Chianelli R.R., Day C.S., Inorg.Chem., 1994, 33, 4418) Диcульфид рения устойчив на воздухе без нагревания, выше 300 оС сгорает, выделяя оксиды рения и серы. Его нагревание до 1000 оС без доступа воздуха приводит к образованию металла, содержащего примесь серы. Такой же продукт образуется при восстановлении дисульфида водородом при 700 оС. Горячая азотная кислота окисляет ReS2 до рениевой кислоты. Известен также и дисульфид технеция, полученный из высшего сульфида нагреванием с серой при 1000 оС, или термолизом.
КОНЕЦ ДОПОЛНЕНИЯ
Химия марганца, технеция и рения в степенях окисления 5, 6, 7
Химия кислородных соединений в высших степенях окисления наиболее разнообразна у технеция и рения, а для марганца она представлена тетраоксоманаганат-ионом MnO4n–, свойства которого зависят от степени окисления атома марганца. Манганаты(V), например, K3MnO4 исторически называют гипоманганатами, манганаты(VI) (K2MnO4) – манганатами, а манганаты(VII) (KMnO4) – перманганатами. Согласно диаграмме вольт-эквивалент-степень окисления (рис.5.2) и значениям стандартных электродных потенциалов, перманганаты устойчивы в кислых, нейтральных и слабощелочных средах, а гипоманганаты и манганаты – в сильнощелочных. Это необходимо учитывать при их синтезе. Ионы MnO4n– представляют собой правильные тетраэдры со значительной долей π-связывания, заключающегося в перекрывании заполненных р-орбиталей атомов кислорода со сходными по симметрии d-орбиталями марганца. Степень π-связывания , а следовательно, и кратность сязи Mn–O возрастает по мере увеличения степени окисления марганца, что приводит к уменьшению длины связи Mn–O от 0.171 нм в MnO43– до 0.166 нм в MnO42– и 0.163 нм в MnO4–. Все оксоманагнаты имеют яркую окраску, зависящую от степени окисления: гипоманганаты – синие, манганаты – зеленые, перманганаты – фиолетовые. Хотя в ионах MnO43– и MnO42– содержатся неспаренные электроны, окраска всех трех типов оксоманаганганатов обусловлена не d-d переходами, а интенсивной полосой переноса заряда. Перманганаты щелочных металлов изоморфны перхлоратам, манганаты – сульфатам и хроматам, а гипоманганаты – ортофосфатам и ванадатам. Эти соли можно использовать в качестве матриц для стабилизации неустойчивых ионов.
Гипоманганаты присутствуют в продуктах высокотемпературарного термического разложения перманганатов или манганатов:
500-850 оС:
3K2MnO4 = 2K3MnO4 MnO2 O2↑,
а также окисления пиролюзита кислородом в сильно щелочной среде:
300 оС
2MnO2 6КОН 1/2О2 = К3MnO4 3Н2О.
Полученные таким образом, они представляют собой сине-зеленые кристаллические порошки, устойчивые только в отсутствии следов влаги из-за легкости диспропорционирования:
2MnO43- 2Н2О = MnO42- MnO2 4ОН—
На сухом воздухе они выдерживают нагревание до 900 оС. В растворе их удается стабилизировать только на холоду и в сильно щелочной среде (константа скорости реакции диспропорционирования составляет 6.9 л/моль–1×с–1 в 3М KOH и > 104 л/моль–1×с–1 при рН = 12 и 20°C). Например, К3MnO4 при –15 оС растворяется в холодном 40 %-ом гидроксиде калия, образуя ярко-синий раствор, из которого могут быть получены кристаллогидраты голубого цвета. Однако уже при 0 оС эти растворы подвергаются диспропорционированию. Небесно-голубые кристаллы соли Na3MnO4×0,25NaOH×12H2O, синтез которой приведен в практических руководствах:
2KMnO4 2Na2SO3 6NaOH 2H2O = 2Na3MnO4 2Na2SO4 2KOH
несмотря на частичную стабилизацию гидроксид-ионами, разлагаются в течение нескольких часов.
Неустойчивые голубые растворы, по-видимому, содержащие гипоманганат-ионы в протонированной форме HMnO42–, удается получить при восстановлении манганата калия в сильно-щелочной среде (10 M KOH) пероксидом водорода или формиатом натрия (J.D. Rush, B.H.J. Bielski, Inorg. Chem., 1995, 34, 5832; D.G. Lee, T. Chen, Journ. Amer. Chem. Soc., 1989, 111, 7535). При этом к охлажденному щелочному раствору манганата восстановитель надо добавлять по каплям. Удобным способом увидеть красивую ярко-голубую окраску гипоманганата является восстановление перманганата в расплавленной щелочи. Этот способ, разработанный одним из авторов книги, до сих пор не описан в литературе, поэтому мы приводим методику эксперимента. В сухую пробирку положите десяток гранул твердого гидроксида калия и добавьте к ним одну каплю раствора перманганата калия. Нагрейте пробирку на пламени. Сначала расплав стает изумрудно-зеленым вследствие образования манганата, а при дальнейшем нагревании – ярко-голубым. При охлаждении он затвердеет, сохраняя окраску. Гипоманганат-ионы оказывают стабилизированными в структуре твердой щелочи.
Разложение безводных гипоманганатов протекает лишь при высоких температурах, и его направление зависит от природы катиона. Так, натриевая соль выше 1250 оС вследствие выделения летучего оксида натрия отщепляет кислород, превращаяясь в MnO
Калийная соль при разложении дает оксоманганаты(III) или смешанную фазу K2Mn4O7,8, в которой марганец находится в двух степенях окисления. На влажном воздухе при 250 оС K3MnO4 окисляется:
2K3MnO4 1/2О2 H2O= 2K2MnO4 2КОН.
Оксоманганат (V) Li3MnO4, полученный в токе кислорода по реакции:
120 оС
LiMnO4 2LiOH = Li3MnO4 H2O 1/2O2↑
при мольном соотношении перманганата калия и гидроксида лития, взятого в форме моногидрата, 1 : 3.1, термически менее устойчив, чем калийная и натриевая соли.
Впервые манганаты(VI)приготовил Глаубер в 1659 г. при растворении пиролюзита в расплавленной селитре. Эти вещества представляют собой темно-зеленые, почти черные кристаллы, растворимые в растворах щелочей с образованием изумрудно-зеленых растворов. Магнитный момент составляет 1,78-1,84 М.Б., что соответствует одному неспаренному электрону d1. В нейтральных и слабокислых растворах манганаты легко диспропорционируют:
3K2MnO4 Н2О = 2KMnO4 MnO2↓ 4КОН,
о чем судят по выпадению черного осадка диоксида и появлению малинового окрашивания. Для ускорения процесса обычно прибегают к пропусканию через раствор углекислого газа:
3K2MnO4 2СО2 = 2KMnO4 MnO2↓ 2К2СО3
или добавляют соль аммония:
3K2MnO4 4NH4Cl = 2KMnO4 MnO2↓ 4NH3 4KCl 2H2O.
Среди манганатов наибольшее значение имеет калийная соль K2MnO4, служащая для производства перманганата KMnO4. Ее синтезируют окислением пиролюзита в присутствии кислорода в расплаве щелочи или в ее концентрированном растворе
MnO2 2KOH 1/2O2 = K2MnO4 H2O.
В качестве окислителей используют также хлорат калия и нитрат калия или их смесь:
3MnO2 6KOH KClO3 = 3K2MnO4 KCl 3H2O,
MnO2 2KOH KNO3 = K2MnO4 KNO2 H2O.
Окисление этими реагентами с заметной скоростью начинается при 170 оС. Продукт извлекают водой, и прозрачный раствор кристаллизуют. При этом часть вещества теряется из-за диспропорционирования.
Этот процесс протекает в несколько стадий. Сначала реакция не сопровождается изменениями степени окисления марганца:
MnO2 2КОН = K2MnO3 Н2О.
Затем, при расплавлении щелочи (380 оС), наступает быстрое окисление MnО2:
2MnO2 6КОН 1/2О2 = K3MnO4 Н2О.
Для проведения последующей стадии требуется понижение температуры ввиду более низкой термической устойчивости манганата по сравнению с гипоманганатом:
K3MnO4 Н2О 1/2О2 = K2MnO4 2КОН.
Максимальный выход K2MnO4 достигается при 190-210 оС. Помимо манганита и гипоманганата, в качестве промежуточных продуктов зафиксированы также K2Mn8O16, K2Mn4O8 и K2Mn2O(4-δ). При синтезе манганата из пиролюзита в жидкой фазе процесс проводят в 40 %-ом растворе КОН при температуре 150-160 оС и давлении кислорода 100 атм. Продукт кристаллизуют и промывают 60 %-ным раствором гидроксида калия при 100 оС.
В электрохимическом методе синтеза пиролюзит используют в качестве анода. Катодом служит железная трубка, электролиз проводят в 90 %-ном КОН при 200 оС. На ряде производств анодному окислению подвергают металлический марганец без примеси железа, при этом образуется смесь перманганата и манганата с выходом 80 %. Повышая температуру процесса и концентрацию щелочи, удается увеличить количество манганата.
В качестве окислителей при синтезе манганатов могут быть использованы пероксиды и азиды:
BaO2 MnO2 = BaMnO4,
4MnO2 KN3 7KOH H2O = 4K2MnO4 3NH3↑.
В лабораторных условиях манганаты щелочных металлов удобно получать окислением воды перманганатом в сильно щелочном раcтворе. Для этого перманганат помещают в концентрированный раствор щелочи и нагревают:
2NaMnO4 2NaОН = 2Na2MnO4 Н2О 1/2О2↑.
В качестве восстановителя может быть использован амид калия в жидком аммиаке:
6KMnO4 6KNH2 = 6K2MnO4 4NH3↑ N2↑.
Манганаты термически менее устойчивы, чем гипоманганаты: K2MnO4 разлагается при 600 оС, выделяя кислород.
Для синтеза манганатов рубия, цезия и бария используют обменные реакции:
Na2MnO4 Ba(OH)2 = BaMnO4↓ 2NaOH.
При увеличении степени окисления марганца термическая устойчивость оксоманганатов уменьшается, а их окислительное действие усиливается, что, например, следует из значений стандартных электродных потенциалов. Практического значения манганаты и гипоманганаты не имеют, так как в водных растворах легко диспропорционируют, давая перманганат-ионы.
Малиново-фиолетовые растворы, содержащие перманганат-ионы MnO4–, также образуются при окислении солей марганца(II) оксидом свинца(IV), висмутатом натрия, персульфатом аммония, иодной кислотой и другими сильными окислителями:
2Mn(NO3)2 5PbO2 6HNO3 = 2HMnO4 5Pb(NO3)2 2 H2O,
2Mn(NO3)2 5NaBiO3 16HNO3 = 2HMnO4 5Bi(NO3)3 5NaNO3 7H2O,
2MnSO4 5(NH4)2S2O8 8H2O = 2HMnO4 5(NH4)2SO4 7H2SO4.
Эти реакции проводят в кислой среде, так как в этих условиях перечисленные выше вещества в наибольшей степени проявляют окислительные свойства.
Для проведения реакций берут очень немного соли марганца, так как в избытке происходит ее сопропорционирование с образующимся перманганатом. Таким образом, эти превращения не могут применяться для синтеза перманганатов, а служат лишь удобным аналитическим методом определения ионов марганца(II). При использовании в качестве восстановителя персульфата требуется присутствие катализатора – ионов Ag . Механизм реакции полностью не изучен, однако известно, что лимитирующей стадией этого процесса является окисления персульфатом ионов серебра, возможно, в форме комплекса
S2O82– 2Ag = 2Ag2 2SO42–
которые и осуществляют последовательное окисление марганца(II) до перманганата. Аналогично персульфату реагирует пероксофосфат P2O84– (Jagan Mohana Rao K., Vittal A.S.P., Krishna Rao P.V. Inorg. Chem., 1984, 23, 3212)
Перманганаты лития, натрия, кальция и стронция сильно гигроскопичны и образуют ряд кристаллогидратов, например, LiMnO4.3H2O, NaMnO4.3H2O, NaMnO4.H2O (Рис.5.35. Фазовая диаграмма системы NaMnO4 – H2O). Растворимость перманганатаов щелочных металлов убывает с ростом катиона, соль лития является исключением (Табл.5.4).
Таблица 5.4. Растворимость некоторых перманганатов (г безводной соли в 100 г воды) при 20 °C
| Катион | Li | Na | K | Rb | Cs | Ca | Sr | Ba | NH4 | Ag |
| Растворимость соли | 32,5 | 59,0 | 6,3 | 1,1 | 0,23 | 62,5* | 7,4** | 0,92 |
* — при 11°C, ** — при 15°C.
Перманганат аммония начинает разлагаться уже при 70°C, а при ударе или нагревании до 110 оС взрывается (L. Kotai, P. Szabo, A. Keszler, Thermochim. Acta, 1999, 338, 129):
2NH4MnO4 = 2MnO2 H2O 3/2O2↑ 2NH3↑.
Перманганат рубидия при нагревании ведет себя так же, как и калийная соль. Разложение гидрата перманганата кальция приводит к отщеплению кристаллизационной воды и последующему разложению соли при 200 °C с общей потерей массы, соответствующей образованию шпинели CaMn2O4. Бариевая соль разлагается при 220 °C.
Синтез перманганатов в лаборатории проводят окислением манганатов хлором, бромом или периодатом в щелочной среде:
2K2MnO4 Cl2 = 2KMnO4 2KCl.
Реакция протекает количественно. Из сильно концентированных растворов сразу выпадают кристаллы. Окисление манганатов в щелочных растворах ионами ClO– или H3IO62– включает диспропорционирование:
2MnO42– = MnO43– MnO4–
и окисление возникающего гипоманганата:
MnO43– H3IO62– = MnO4– IO3– 3OH—.
Окисление озоном не находит практического применения:
2K2MnO4 О3 Н2О = 2KMnO4 2КОН О2↑.
Другими способами являются диспропорционирование манганатов, а также обменные реакции между солями:
KMnO4 NH4Cl = NH4MnO4↓ KCl (осаждается при охлаждении),
AgMnO4 NaCl = NaMnO4 AgCl↓.
Таким же способом удается получить и растворы марганцевой кислoты НМnO4:
Ba(MnO4)2 H2SO4 = BaSO4↓ 2HMnO4,
2KMnO4 H2SiF6 = K2SiF6↓ 2HMnO4.
Марганцевая кислота относится к сильным кислотам, поэтому перманганаты щелочных металлов практически не гидролизуются. Разбавленные свежеприготовленные водные растворы марганцевой кислоты можно нагревать до кипения без заметного разложения. Однако, при длительном хранении водные растворы постепенно разлагаются с образованием осадка MnO2 и выделением кислорода:
4НМnO4 = 4MnO2↓ 3О2↑ 2Н2О.
Марганцевая кислота известна только в водных растворах.
Выпариванием получен насыщенный при 18 оС раствор с концентрацией 1,4 моль/литр (17 г/100 мл раствора). Растворением марганцевого ангидрида в воде достигнуты концентрации кислоты до 20 %. Полное разложение 0,3 М раствора при комнатной температуре происходит за два месяца. При попытках выделения твердой кислоты вымораживанием растворов получены кристаллы (H3O)2[Mn(MnO4)6].11H2O, в состав которых входят атомы марганца в разных степенях окисления, Mn(IV) и Mn(VII) (B.Krebs, K.D.Hasse, Angew. Chem., 1974, 86, 647). Имеются сведения о выделении кристаллов безводной кислоты HMnO4 и ее оксониевой соли H5O2 MnO4–.
Соли марганцевой кислоты – одни из важнейших неорганических окислителей, активные в твердой фазе и лабильные в растворе. С их помощью можно окислить многие органические и неорганические вещества. Такой широкий спектр действия объясняется сложным механизмом окиления, включающим несколько параллельных путей, хотя бы один из которых практически всегда оказывается возможным, а высокая скорость протекания процессов обусловлена низкой энергией активации – от 21 до 42 кДж/моль. Наиболее сильными окислительными свойствами перманганаты обладают в кислотной среде, где они восстанaвливаются до ионов Mn2 :
MnO4– 8H 5e– ® Mn2 4H2O, E0 = 1,51 B (pH = 0).
В нейтральной и слабощелочной среде перманганат восстанавливается до диоксида марганца, выпадающего из раствора в виде бурого осадка
MnO4– 2H2O 3e– ® MnO2 4OH–, E0 = 1,15 B (pH = 7).
а в сильнощелочной (рН > 12,5) образуется изумрудно-зеленый манганат, устойчивый при рН > 14:
MnO4– e– ® MnO42– , E0 = 0,56 B (pH = 14).
Хотя механизмы многих реакций с участием перманганат-иона являлись предметом тщательного изучения, однозначного вывода о протекании процессов сделать не удается. Механизм восстановления перманганат-иона включает несколько стадий, представляющих собой одно- и двухэлектронные переносы. В качестве интермедиата в щелочной среде практически всегда удается зафиксировать гипоманганат, дающий сигналы в спектре электронного парамагнитного резонанса и характерный электронный спектр (Рис.5.36. Электронные спектры перманагната калия и продуктов его восстановления, включая промежуточные соединения; 20 °C, 0.2 ммоль/литр). Приведем предполагаемый механизм реакции взаимодействия перманганата с сульфитом при рН = 9 (L.I. Simandi, M. Jaki, C.R. Savage, Z.A. Schelly, Journ. Amer. Chem. Soc., 1985, 107, 4220; 1984, 106, 6866). Первой стадией процесса являются два параллельных внешнесферных одноэлектронных переноса, приводящих к окиcлению сульфит-иона до сульфата:
MnO4– SO32– ® MnO42– SO3–
MnO4– SO3– 2OH– ® MnO42– SO42– H2O.
Образующийся манганат-ион является более слабым и более медленно действующим окислителем, чем перманганат, поэтому он успевает претерпеть диспропорционирование, которое приводит к гипоманганату:
2MnO42–® MnO4– MnO43–
Гипоманганат, в свою очередь, также диспропорционирует на перманганат и диоксид марганца, который сначала образует коллоидный раствор желтого цвета, а лишь спустя некоторое время осаждается:
2MnO43–® MnO2(sol) MnO4–,
MnO2(sol) ®MnO2¯.
Суммарное уравнение имеет вид:
2MnO4– 3SO32– H2O ® 2MnO2¯ 3SO42– 2OH–.
Восстановление перманганатом сульфита в кислой среде протекает через интермедиаты — соединения марганца(III). Их удалось стабилизировать в виде K2[MnF3SO4]и K2[MnF5], введя в раствор фторид-ионы (C. Bhattacharyya et al, Journ. Chem. Soc., Dalton Trans., 1993, 4397).
С органическими веществами, например, спиртами или олефинами, перманганат-ион взаимодействует по внутрисферному механизму через хелатный цикл, внутри которого и происходит двухэлектронный перенос. Дальнейший механизм определяется рН раствора. В сильнощелочной среде манганат(V) достаточно устойчив к диспропорционированию, поэтому окисление происходит под действием другого перманганат-иона. В среднещелочных, нейтральных и кислотных растворах протекает диспропорционирование. Конечный продукт реакции, диол, образуется в результате гидролиза хелатного комплекса, а манганат-ионы, диспропорционируя, переходят в диоксид марганца ().D. G. Lee, T. Chen, Journ. Amer. Chem. Soc., 1989, 111, 7534).
К недостаткам перманганатов как окислителей следует отнести невозможность их использования в органических растворителях, по причине их низкой растворимости в малополярной среде. Однако это удается преодолеть при использовании межфазного катализа, например, добавляя краун-эфиры (см. т.2, стр 46).
ДОПОЛНЕНИЕ. Перманганат калия.
Перманганат калия представляет собой фиолетово-черные блестящие гигроскопичные кристаллы, растворимые в воде с образованием ярко окрашенных малиновых растворов. Насыщенный раствор этой соли (растворимость в 100 г воды: 6,4 г при 20°C, 22,2 г при 60°C, Рис.5.37. Кривая растворимости перманганата калия в воде; координаты эвтектики: 2,91%, –0,58°C) почти черного цвета. В кристаллическом перманганате KMnO4 атомы марганца располагаются в правильных тетраэдрах MnO4, в которых межатомные расстояния составляют: Mn-O 0,162 – 0,163 нм, а углы О-Mn-O 108,9-111,0 о. Связи Mn-O ковалентные. Перманганат калия изоморфен с перманганатами рубидия, цезия, аммония, а также с перхлоратами калия и таллия(I), сульфатами бария и стронция, хроматом бария, метапериодатом и тетрафтороборатом калия. Интересно, что он не образует смешанных кристаллов с перманганатом натрия. Недавно получены смешанные кристаллы манганата-перманганата калия K3Mn2O8 (M.B. Hursthouse et al, Journ. Chem. Soc., Faraday Trans., 1992, 88, 3071 ).
При нагревании перманганат разлагается с выделением кислорода:
220 °C
2KMnO4 = K2MnO4 MnO2 О2↑.
Дальнейший ход разложения представлен на рис.5.38 (Рис.5.38 Кривая потери массы при разложении перманганата калия на воздухе, скорость нагрева 10 °/мин, материал тигля — алунд. Метод изучения изменения массы вещества при нагревании, называют термогравиметрическим анализом, а представленную кривую – дериватограммой).
Выше 100 оС он восстанавливается водородом:
10KMnO4 10Н2 = 5K2MnO4 5MnO2 10H2O.
Смеси KМnO4 с серой, фосфором, мышьяком, сурьмой, порошками кремния, алюминия, титана при растирании или нагревании загораются, при соприкосновении кристаллов перманганата с жидкой серой происходит взрыв. Смесь тонкого порошка углерода с перманганатом может служить для зажигания. Все это свидетельствует о высокой окислительной активности перманганата. В водных растворах он легко окисляет сульфиты, сульфиды, иодиды, оксалаты и многие другие типичные восстановители, а также органические вещества, содержащие связи С=С, СºС, С–ОН, С=О.
При растворении перманганата калия в олеуме образуется зеленый раствор
KMnO4 3H2S2O7 = K MnO3 HSO4– HS3O10– 2H2SO4,
из которого через 2-3 часа выделяется кислород и выпадает MnO2. Раствор KMnO4 в азотной кислоте имеет интенсивный красный цвет, и из него также постепенно выделяется осадок диоксида. Этим пользуются для получения чистого пиролюзита. При обработке перманганата фосфорной кислотой (95-100 %) выделяется Mn2O7. С безводной HF и фторосерной кислотой HSO3F перманганат дает зеленый раствор и пары зеленого цвета, из которых выделен зеленый MnO3F. Интересно, что перманганат калия не растворим в жидком хлороводороде, хотя газообразный HCl или его водный раствор легко окисляются им до свободного хлора.
При 40 оС пентафторид иода взаимодействует с KMnO4 как фторирующий агент:
KMnO4 IF5 = MnO3F IOF3 KF.
Перманганат катализирует термическое разложение хлората калия и перхлората аммония. Благодаря сильным окислительным свойствам, он используется как окислитель при подготовке и очистке воды и воздуха, служит эффективным антисептиком в фармакологии, как реагент в аналитической и органической химии.
Перманганат калия синтезируют электролитическим окислением манганата K2MnO4 в щелочных растворах или анодным окислением марганца и его сплавов:
2K2MnO4 2H2O = 2 KMnO4 2KOH H2↑.
Для устранения катодного восстановления MnO2 процесс проводят с диафрагмой. Исходный раствор содержит 150-250 г/литр K2MnO4 и 100-250 г/литр КОН. За время 10-40 часов при 30-65 оС окисляется 70-95 % K2MnO4. Аналогично протекает анодное окисление манганата натрия Na2MnO4 до перманганата.
Анодное окисление ферромарганца протекает по уравнению:
Mn OН— 3Н2О = MnO4— 3,5 Н2↑.
В процессе электролиза перманганат-ионы образуются на аноде.
При использовании разбавленных растворов щелочи или поташа происходит образование перманганата калия, тогда как при высоких концентрациях щелочи выделяется манганат K2MnO4. При охлаждении раствора с рН 7,5 –10, насыщенного при 60 оС, выпадают кристаллы KMnO4 размером до 4 мм. Всегда присутствующий в водных щелочных растворах перманганата диоксид марганца коагулируют введением до 1 % поверхностно активного вещества и тем самым стабилизируют эти растворы. Аналитически чистый продукт получают двойной перекристаллизацией.
§
КОНЕЦ ДОПОЛНЕНИЯ
При действии на подкисленный раствор перманганата калия различных восстановителей происходит его обесцвечивание:
10FeSO4 2KMnO4 8H2SO4 = 5Fe2(SO4)3 2MnSO4 K2SO4 8H2O,
5H2O2 2KMnO4 3H2SO4 = 2MnSO4 5O2 K2SO4 8H2O,
5H2C2O4 2KMnO4 8H2SO4 = 2MnSO4 K2SO4 10CO2 8H2O.
Реакция окисления пероксида водорода является колебательной и протекает с участием свободных радикалов. В присутствии фосфорной кислоты она совпровождается периодическими изменениями окраски от розовой фиолетовой (Nagy A., Triendl L., Nature, 1986, 320, 344). Взаимодействие перманганата калия с щавелевой кислотой используется в титриметрии. Эта реакция может быть условно разделена на две стадии, различающиеся по скорости протекания и характеру процессов. На первой стадии происходит восстановление перманганат-иона до промежуточного оксалатного комплекса марганца (III):
2MnO4– 4Mn2 8H = 5 Mn(III) 4H2O,
где Mn(III) означает оксалатный комплекс марганца(III), возможно, [Mn(C2O4)3]3–.
Интересно, что этот процесс осуществим с заметной скоростью лишь в присутствии ионов Mn2 , которые первоначально могут содержаться в небольшом количестве в виде примеси в перманганате, а впоследствии образуются на второй стадии процесса. Таким образом, скорость протекания первой стадии существенно возрастает по мере прохождения реакции за счет того, что один из продуктов реакции выступает в качестве реагента – такие процессы называют автокаталитическими. Если перед началом реакции к исходному раствору перманганата калия добавить избыток фторид-ионов, связывающих ионы марганца(II) во фторидный комплекс, то при добавлении щавелевой кислоты обесцвечивание не наблюдается – реакция практически не протекает.
Вторая стадия заключается в восстановлении комплекса марганца(III) и окислении оксалата до углекислого газа:
2Mn(III) C2O42– = 2Mn2 2CO2.
Однозначной трактовки этого процесса в литературе пока не достигнуто. Известно, что разрыв связи С–С в оксалат-ионе, приводящий к образованию углекислого газа, происходит гомолитически, то есть по радикальному механизму, однако состав образующихся радикалов (ими могут быть С2О4–× , СО2–×) точно не установлен (J.M. Malcolm, R.M. Noyes, Journ. Phys. Chem., 1952, 74, 2769; R.T. Powell, T. Oskin, N. Ganapathisubramanian, Journ. Phys. Chem., 1989, 93, 2718; V.Pimienta, D. Lavabre, G. Levy, J.C. Micheau, Journ. Phys. Chem., 1995, 99, 14365).
В степени окисления 7 для технеция и рения известны соединения, аналогичные соединениям марганца, однако, в отличие от перманганатов, они являются слабыми окислителями. Большие атомные радиусы обусловливают существование оксо-анионов с координационными числами пять и шесть, напоминающими соответствующие периодаты.
При взаимодействии высших оксидов с водой образуются растворы технециевой НТсО4 и рениевой HReO4 кислот. Препаративными методами синтеза служат также окисление металлов или диоксидов пероксидом водорода
2ТсО2 3Н2О2 = 2НТсО4 2Н2О,
азотной кислотой:
Re 7HNO3 = HReO4 3H2O 7NO2↑,
хлорной водой:
2ReO2 3Cl2 4H2O = 2HReO4 6HCl..
Медленное испарение водного раствора технециевой кислоты приводит к темно-красным кристаллам состава НТсО4 неизвестного строения. Из растворов рениевой кислоты выделены желтоватые кристаллы Re2O7.2H2O, представляющие собой гидратированную молекулу рениевого ангидрида, состоящую из тетраэдра [ReO4] и октаэдра [ReO6], соединенных общей вершиной (Beyer H., Glemser O., Krebs B., Wagner G., Z. Anorg. Allg. Chem., 1970, 375, 87), а также гидрат HReO4.H2O, построенный из тетраэдрических молекул HReO4, между которыми находятся молекулы воды, удерживаемые системой водородных связей (Wlschek G., Svoboda I., Fuess H., Z. Anorg. Allg. Chem., 1993, 619, 1679) (Рис.5.39. Строение гидратов (а) молекула Re2O7.2H2O, (б) кристаллическая решетка HReO4.H2O) Кристаллы безводной кислоты HReO4 не получены. Молекулы HReO4 устойчивы в парообразном состоянии, при кипячении концентрированных растворов кислота частично улетучивается.
Технециевая и рениевая кислоты, подобно марганцевой, представляют собой сильные одноосновные кислоты: их константы диссциации в неводных растворителях (рКа от –8 до –9, HReO4 несколько сильнее HTcO4) сравнимы с HBr, в разбавленных водных растворах они полностью диссоциированы.
С активными металлами (Mg, Zn, Fe) кислоты реагируют с выделением водорода, а при взаимодействии с оксидами, гидроксидами, карбонатами образуют соли. Пертехнетаты и перренаты многих металлов устойчивы и выделены в кристаллическом виде. Известно несколько типов солей, образованных тетраэдрическими анионами МO4– (например, NaReO4 метаперренат натрия), анионами МO53– с формой тригональной бипирамиды или квадратной пирамиды (Na3ReO5 мезоперренат натрия) и октаэдрическими МO65– (Na5ReO6 ортоперренат натрия), которые часто изоструктурны ортопериодатам. Мезо- и орто-формы солей, подобно аналогичным периодатам, получают не из растворов, а сплавлением с оксидами металлов:
1000 oC
Ba(ReO4)2 4BaCO3 = Ba5(ReO6)2 4CO2,
500 oC
NaTcO4 Na2O = Na3TcO5
Спектры поглощения всех трех ионов MO4– (M = Mn, Tc, Re) очень близки и обусловлены переходом электрона с орбиталей, локализованных на атомах кислорода, на орбитали металла. Интенсивная окраска иона перманганата вызвана тем, что полоса переноса заряда частично попадает в видимую области спектра, а пертехнетата и перрената она полностью находится в ультрафиолете, поэтому они бесцветные. Интересно, что полоса поглощения пертехнетат-иона располагается очень близко к краю видимой области. Поэтому даже небольшие искажения тетраэдрического иона TcO4—, которые неизбежно существуют при тесном взаимодействии ионов в кристаллической решетке или концентрированном растворе, приводят к тому, что край полосы захватывает фиолетовую часть видимой области, этим и объясняется красный цвет кристаллической кислоты НТсО4 и красно-коричневая окраска некоторых ее солей (Рис.5.40. Строение перренатов натрия: (а) NaReO4, (б) Na3ReO5, (в) Na5ReO6).
В отличие от иона перманганата, они устойчивы в щелочных растворах и характеризуется слабыми окислительными свойствами. Так, при действии на подкисленные растворы пертехнетатов и перренатов сероводорода выпадают черные осадки высших сульфидов:
2NaReO4 H2SO4 7H2S = Re2S7¯ Na2SO4 8H2O.
Для препаративного получения сульфидов в качестве восстановителя удобно использовать тиосульфат натрия Na2S2O3:
2KReO4 7Na2S2O3 H2SO4 = Re2S7↓ 7Na2SO4 K2SO4 H2O.
Гептасульфиды плохо растворим не только в воде, но и неокисляющих кислотах и щелочах. Азотная кислота переводит их в технециевую и рениевой кислоты. При нагревании на воздухе сульфиды сгорают:
2Re2S7 21O2 = 2Re2О7 14SO2,
а при нагревании в вакууме термически диссоциирует на дисульфид ReS2 и серу. Водород при 1100 оС восстанавливает их до металла. При взаимодействии сульфида рения(VII) c сульфидами щелочных металлов образуются тиоперренаты, например, NaReS4. С алкенами и алкинами они дают продукты присоединения. Cмешанные тио-оксо соли примерного состава NaReO3S обусловливают желтое окрашивание нейтрального или щелочного раствора перрената при пропускании через него сероводорода.
Окислительные свойства пертехнатов и перренатов в водных растворах проявляются лишь в кислой среде под действием сильных восстановителей, например, иодидов, солей олова(II), титана(III), гипофосфитов:
2KReO4 6KI 16HCl = 2K2ReCl6 4KCl 3I2 8H2O,
2NH4ReO4 2NH4Cl 10HCl 3H3PO2 = 2(NH4)2ReCl6 3H3PO3 5H2O,
2HReO4 28HCl 7SnCl2 = 2Re 7H2SnCl6 8H2O.
Получены пертехнетаты щелочных, щелочноземельных и редкоземельных металлов, аммония, таллия(I) и серебра(I). Белый пертехнетат калия КТсО4 выделяется в виде бесцветных кристаллов при взаимодействии пертехнетата аммония с едким кали, он плавится при 540 оС и возгоняется без разложения около 1000 оС. При нагревании в водороде КТсО4 восстанавливается до металла, с монооксидом углерода образует карбонил Тс2(СО)10. Взаимодействием его водных растворов с сильными восстановителями (Sn2 , Cr2 ) в присутствии лигандов получают комплексные соединения технеция(IV).
Среди пертехнетатов наиболее важен пертехнетат аммония NH4ТсО4– исходное соединение для получения всех остальных производных технеция. Он не гигроскопичен, в вакууме при 500 оС разлагается до ТсО2. Очень чистое соединение может быть возогнано без разложения. Растворение NH4TcO4 в жидком фтороводороде приводит к оксофториду TcO3F, взаимодействие с хлористым тионилом – к оксохлориду (NH4)2TcO2Cl4. Растворимость пертехнетатов щелочных металлов в воде понижается с ростом размера катиона (растворимость CsTcO4 при 20°C 0,412 г/100 г раствора); наименее растворим пертехнетат таллия(I) (0,072 г/100 г раствора ). Пертехнетаты, в отличие от перренатов, обладают сильными антикоррозионными свойствами даже при весьма небольших концентрациях в растворах (10–5 М). Механизм ингибирования коррозии включает обратимую адсорбцию иона на дефектах поверхности и связан с окислительно- восстановительными свойствами пары ТсО4– – ТсО2.
Перренаты щелочных металлов, аммония, таллия(I) и серебра(I) устойчивых гидратов не образуют. Растворимость в воде солей калия и аммония характеризуются сильной температурной зависимостью (от 0 до 50 оС растворимость КReO4 возрастает в 9 раз, а NH4ReO4 – в 5 раз), это позволяет очищать их методом перекристаллизации. Как и у пертехнетатов, наименее растворимо соединение таллия TlIReO4 (0,113 г/100 г раствора). Перренат калия плавится без разложения при 555 оС. Перренат аммония при 200 оС на воздухе разлагается с частичным испарением летучего оксида Re2O7 и выделением черного диоксида ReO2. Его распад в инертной атмосфере сопровождается образованием исключительно диоксида. Водородом перренат аммония восстанавливается до металла. Известны также перренаты некоторых двух- (Ca, Sr, Ba, Cu, Zn, Cd, Pb) и трехзарядных (Al, РЗЭ) катионов. Из водных растворов все они кристаллизуются в виде гидратов.
Мезопертехнетаты и перренаты щелочных металлов – вещества оранжево-коричневого цвета. Они характеризуются высокой гигроскопичностью и сравнительно низкой термической устойчивостью, часто они изоструктурны аналогичными соединениями иода.
При сопропорционировании пертехнетатов и перренатов с металлами или диоксидами получены соединения в степени окисления 5:
500 °C
2TcO2 Na2O NaTcO4 = 3NaTcO3,
LiTcO4 Tc Li2O = Li3TcO4
При 1000 оС они диспропорционируют:
11Li3TcO4 = 8Li4TcO5 2Tc LiTcO4.
Химия комплексных соединений технеция и рения в высшей степени окисления ограничена всего несколькими соединениями. Выделен комплекс H[ReO2(SO4)2].3H2O, выпадающий из концентрированных сернокислых растворов рениевой кислоты. Достаточно редко ион перрената выступает в качестве лиганда, как, например, в катионе уранила [UO2ReO4] и соединении UO2ReO4.
Для рения( 5) известны комплексные соединения, например, цианиды K3[Re(CN)8], K3[ReO2(CN)4], K3Na[Re2O3(CN)8]. Последний представляет собой димер, [(CN)4(O)Re–O–Re(O)(CN)4]4– (Lumme P.O., Turpeinen U., Stasicka Z., Acta Cryst. C., 1991, 47, 501).
Оксосоли технеция(VI), подобно пертехнетатам, существуют в мета- (Li2ТсО4), мезо- (Li4TcO5) и ортоформах (Li6TcO6). Фиолетово-синий технетат К2ТсО4 получают восстановлением раствора пертехнетата КТсО4 на ртутном катоде. Мезо- и ортоформы образуются при твердофазных реакциях, аналогичных синтезу технетатов(V):
6LiTcO4 Tc 11Li2O = 7Li4TcO5
6LiTcO4 Tc 18Li2O = 7Li6TcO6.
В воде все эти соединения неустойчивы вследствие диспропорционирования:
3ТсО42– 2Н2О = 2ТсО4– ТсО2↓ 4ОН–.
В отличие от марганца и технеция, у рения выделена ренистая кислота H2ReO4 и ее соли – ренаты. Они образуются при взаимодействии триоксида рения с концентрированными растворами щелочей. Зеленая окраска этих растворов обусловлена анионами ReO42–. Ренаты щелочных металлов при растворении в воде диспропорционируют:
2K2ReO4 2H2O = 2KReO4 ReO2↓ 4KOH.
Гидрат триоксида, ренистая кислота H2ReO4, образуется в виде черного аморфного порошка при окислении рения кислородом в присутствии паров воды. Она нерастворима в воде, слабо растворима в растворах кислот. С горячими растворами щелочей она дает ренаты. Азотная кислота, пероксид водорода и другие окислители переводят ренистую кислоту в рениевую.
Орто-формы ренатов получают твердофазными реакциями:
3Ca(ReO4)2 Re 18CaO = 7Ca3ReO6
Кристаллические структуры ренатов неизвестны.
§
Среди металлов седьмой группы лишь марганец образует бинарные соединения с водородом, они имеют переменный состав и мало изучены. Весьма интересные и необычные вещества представляют собой ионные гидриды технеция и рения, содержащие атомы металла в высшей степени окисления. Они не имеют аналогов в химии других элементов. Бесцветные диамагнитные кристаллы нонагидридорената(VII) натрия Na2ReН9 получают восстановлением перрената металлическим натрием в этаноле без доступа воздуха:
NaReO4 18Na 13C2H5OH = Na2ReН9 4NaOH 13C2H5ONa.
Это вещество растворимо в воде, но устойчиво в растворе лишь в присутствии щелочи.
Строение комплексного иона соответствует тригональной призме с тремя атомами водорода над каждой прямоугольной гранью (Рис.5.41. Строение гидридных комплексов рения: (а) ReН92–, (б) димер H6Re2(μ-H)3PR3, PR3 = 1,1,1-трисдифосфинометилэтан; у молекулы фосфина показаны только атомы фосфора). Расстояния между атомами Re и призматическими Н равны 0,168 нм. Несмотря на различные позиции призматических и шапочных атомов водорода, в спектре ПМР присутствует только один сигнал, показывающий равноценность всех атомов Н и обусловленный их быстрым обменом между разными позициями. Связи Re–H в гидридных комплексах ковалентные, так как образуются в результате перекрывания заполненных орбиталей гидрид-иона с вакантными d-орбиталями рения.
При восстановлении перрената калия, меченого радиоактивным изотопом 99Тс, раствором металлического калия в этилендиамине было замечено, что весь введенный радионуклид оказывается сосредоточенным в осадке комплексного гидрида К2ReН9. Отсюда сделали заключение о существовании аналогичного соединения у технеция. Позже его получили из пертехнетата аммония по методике, разработанной для рениевого комплекса. Связи Тс-Н слабее связей Re-Н, что соответствует большей реакционной способности технециевого соединения. Это соответствует увеличению стабильности высших степеней окисления при переходе вниз по группе. Благодаря наличию водорода в степени окисления –1, гидридные комплексы технеция и рения являются восстановителями, причем у К2TcН9 эти свойства проявляются сильнее – он восстанавливает нитрат таллия сразу до металла без образования белого осадка соединения таллия(I), наблюдаемого при использовании К2ReН9. Помимо чисто гидридных комплексов, синтезированы гидриды со смешанными анионами, например, c замещенными фосфинами ReH7PR3, а также двухъядерные кластеры, в которых фрагмент RePR3 соединен с ионом [ReH9]2- через три мостиковых гидрида и посредством тройной связи Re≡Re (Рис. б) (S.C. Abrahams, A.P. Ginsberg, T.F. Koetzle, P. Marsh, C.R. Sprinkle, Inorg. Chem., 1986, 25, 2500).
Известны также комплексные гидриды рения в степени окисления 3, например, гексагидридоренат(III) калия K3ReH6 (Bronger W., Auffermann G., Schilder H., Z. Anorg. Allg. Chem., 1998, 624, 497).
КОНЕЦ ДОПОЛНЕНИЯ
§
Перед описанием свойств важнейших соединений, сформулируем важнейшие особенности химии галогенидов. По мере увеличения степени окисления элемента возрастает ковалентный характер соединений, что сказывается в усилении гидролиза, понижении температур плавления. Это приводит также к росту окислительной ативности атомов металла, в том числе во внутримолекулярных окислительно-восстановительных реакциях, что обусловливает понижение термической устойчивости галогенидов, например, в ряду MnCl2 – MnCl3 – MnCl4. Для переходных металлов характерно образование комплексов, в том числе и галогенидных, которые часто оказываются более устойчивыми, чем бинарные галогениды. Окислительная активность галогенидов вниз по группе убывает, что согласуется с общим характером изменения устойчивости степени окисления по группе. В целом при переходе от марганца к технецию и рению понижается устойчивость низших галогенидов и возрастает стабильность высших. Галогениды в низшей степени окисления известны только для марганца, а единственный гептагалогенид является фторидом рения ReF7 (табл.5.5). Высший фторид технеция имеет формулу ТсF6, а марганца — MnF4. Это отображает повышение устойчивости высших степеней окисления при переходе от марганца к рению и одновременно ослабление способности марганца достигать высших степенй окисления по сравнению с соседним элементом хромом, у которого образуются фториды CrF5 и CrF6.
Таблица 5.5. Галогениды металлов седьмой группы
| Степень окисления | Марганец | Технеций | Рений |
| 2 | MnF2, MnCl2, MnBr2, MnI2 | ||
| 3 | MnF3 , MnCl3 | Re3Cl9, Re3Br9, Re3I9 | |
| 4 | MnF4 , MnCl4 | TcCl4 | ReF4, ReCl4, ReBr4, ReI4 |
| 5 | TcF5 | ReF5, Re2Cl10, ReBr5 | |
| 6 | TcF6 | ReF6, ReCl6 | |
| 7 | ReF7 |
Как видно из таблицы, среди всех галогенидов наиболее многочисленны фториды, что объясняется значительной долей ионности. Благодаря этому, часто они имеют структуру, отличную от других галогенидов.
Среди галогенидов марганца наиболее устойчивы соединения со степенью окисления 2. Все они представляют собой бесцветные (в безводном виде) или бледно-розовые (кристаллогидраты) кристаллические порошки, хорошо растворимые в воде (Сноска: MnF2 сравнительно плохо растворим в воде: 1,0 г в 100 г раствора при 24 °C) термически устойчивы, в газообразном состоянии состоят из линейных молекул, из водных растворов выделяются в виде кристаллогидратов. В разбавленных водных растворах они присутствуют в форме гексаакваионов. Однако по мере увеличения концентрации раствора галогенид-ионы начинают входить в координационную сферу марганца:
Mn2 (aq) Cl–(aq)  MnCl , K= 3.85.
MnCl , K= 3.85.
В растворах хлорида марганца появление хлоридных комплексов зафиксировано при концентрации Cl– 0,5 моль/л. В более крепких хлоридных растворах преобладают ионы Mn(Н2О)5Cl (0,36 моль/л), молекулы Mn(Н2О)4Cl2 (~2 моль/л), а при еще большей концентрации хлорид-ионов – октаэдрические анионы MnCl3(H2O)3—, MnCl4(Н2О)22-, MnCl62-. В сильнокислой среде существуют тетраэдрические анионы MnCl42-, а также смешанные аквахлорокомплексы MnCln(H2O)4-n, n = 1 – 3, однако твердые тетрахлороманганаты(II) удается получить лишь сплавлением. При растворении таких соединений в воде или спирте анионы MnCl42- разрушаются. Склонность к образованию галогенидных комплексов возрастает при переходе от фторидов к иодидам.
Спекание с фторидами щелочных металлов приводит к тведым комплексным соединениям NaMnF3, KMnF3, RbMnF3. В них атомы марганца октаэдрически окружены атомами фтора, при этом изолированные полиэдры MnF64- необразуются: регулярные октаэдры MnF6 в этих комплексах соединены между собой всеми вершинами. Также выделены твердые фтороманганаты(II) других составов, например, NaMn3F7, K2MnF4, K3Mn2F7, MII(MnF3)2 (MII-щелочноземельные металлы, кроме Ве).
При нагревании на воздухе дигалогениды окисляются:
3MnF2 1∕2О2 3Н2О = Mn3О4 6НF↑,
2MnBr2 1,5O2 = Mn2O3 2Br2↑.
Фторид марганца(II) при взаимодействии с фтором количественно переводится в MnF3 при 250 оС и в MnF4 при 550 оС. При реакциях MnF2 с фторидами ксенона образуются фтороманганаты(IV):
120 оС
MnF2 2ХеF2 = ХеMnF6 Хе↑,
60 оС
MnF2 5ХеF6 = Хе4MnF28 Хе↑ F2↑.
С активными восстановителями (натрием, цинком) безводные галогениды реагируют с взрывом. Восстановление водородом протекает более спокойно. При нагревании расплава хлорида марганца(II) в атмосфере водорода до 940 оС выделяется металлический марганец в форме тонких игольчатых кристаллов.
В безводном эфире действием на MnCl2 борогидрида лития образуется бесцветный раствор Mn(ВН4)2:
MnCl2 2LiBH4 = Mn(BH4)2 2LiCl↓
Дифторид MnF2, впервые полученный Берцелиусом (1824 г.), нельзя синтезировать прямой реакцией простых веществ. Его получают по реакции карбоната марганца с 40 %-ой плавиковой кислотой, а безводную соль – растворением MnСО3 в избытке расплава NH4HF2
700 оС
MnCO3 NH4HF2 = MnF2 CO2↑ NH3↑ H2O.
Хлорид, бромид и иодид получают галогенированием простых веществ, оксидов, гидратов и других соединений с разнообразными хлорирующими агентами
400 оС
MnO CCl4 = MnCl2 COCl2
Mn2O3 2AlI3 = 2MnI2 Al2O3 I2↑.
Помимо этого, безводные галогениды образуются и при обезвоживании кристаллогидратов. Во избежание гидролиза процесс проводят в токе галогеноводорода или используют соль аммония:
MnCl2.4H2O NН4Cl = MnCl2 4H2O↑ HCl↑ NH3↑.
Среди высших галогенидов марганца хорошо изучены три- и тетрафторид. Относительно существования высших хлоридов MnCl3 и MnCl4 споры среди химиков ведутся уже около 300 лет, но вопрос до сих пор не решен окончательно, а бромиды и иодиды – вообще неизвестны.
Существование трихлорида MnCl3 окончательно не доказано – возможно, это комплекс MnIIи MnIV. Он присутствует в темном растворе, образующемся при растворении пиролюзита в соляной кислоте, а также содержится в твердом неустойчивом осадке, выпадающем при высаливании четыреххлористым углеродом из раствора, полученного взаимодействием эфирной суспензии MnО2 газообразным HCl при –63 оС. Описаны и другие методы получения растворов этого соединения (R. Uson et al, Transition Metal Chem., 1976, 1, 122), среди них — взаимодействие гидроксида марганца(III) с сильно охлажденной конц. HCl или обработка ацетата MnIII сухим жидким хлороводородом при – 100 оС.
Хлорид марганца(III) легко растворяется в жидком хлороводороде, но мгновенно разрушается водой, где для его стабилизации необходима высокая концентрация хлоридных-ионов. Хлороманганаты(III) стабилизируются кристаллической решеткой: выделены кристаллы, Rb2MnCl5.H2O, Cs2MnCl5, содержащие квадратно-пирамидальные (K2MnCl5.H2O) и октаэдрические ([Co(pn)3]MnCl6, pn – 1,2-пропандиамин) ионы.
Тетрахлорид марганца MnCl4, вероятно, существует наряду с трихлоридом в эфирном растворе при действии на MnO2 или перманганат калия соляной кислоты, но крайне неустойчив и распадается с выделением хлора. Об образовании этого вещества свидетельствует факт образования гексахлороманганат(IV) ионов, которые легко могут быть выделены в виде соли при введении в раствор ионов калия:
Ca(MnO4)2 4KCl 16HCl = 2K2MnCl6 CaCl2 8H2O 3Cl2↑.
Эти темно-красные кристаллы неустойчивы даже на сухом воздухе и постепенно разлагаются, выделяя хлор.
Красно-пурпурный трифторид MnF3 получают из дифторида действием фтора или трифторида кобальта:
300 оС
MnF2 СоF3 = MnF3 СоF2,
взаимодействием оксида марганца(III) с газообразным HF при 150 оС. В виде красного осадка MnF3 выделяется при растворении MnCl2 в BrF3.
Кристаллическая решетка MnF3 состоит из соединенных общими вершинами октаэдров MnF6 (Mn-F 0,179 – 0,209 нм), искаженных вследствие эффекта Яна-Теллера. С водой трифторид дает осадок темного цвета и красный раствор, разлагающийся на MnF2, НF и оксигидрат марганца. Выделен его тригидрат MnF3.3Н2О, структура которого состоит из октаэдров MnF63- и Mn(Н2О)63 (Рис. 5.42. Строение MnF3.3Н2О, мне надо сделать рисунок). Тригидрат полностью обезвоживается с помощью конц. H2SO4. При нагревании твердый MnF3 сублимируется, параллельно разлагаясь на дифторид и фтор. Во время нагревания MnF3 в токе фтора при 400 оС образуется тетрафторид MnF4. С фторидом ксенона ХеF2 под давлением образуется ХеMnF6, а в сухой атмосфере при 5 оС с жидким N2O5 образует нитрат Mn(NO3)3 и комплекс NO2[Mn(NO3)4].
Трифторид MnF3 часто служит фторирующим агентом. С его помощью пентахлорид PCl5 переводят в PF5, получают различные фторопроизводные фуллеренов, фторируют различные органические соединения.
Трифторид в растворах и в твердом состоянии образует множество комплексных ионов. Выделены многочисленные твердые фтороманганаты(III), например: Li2MnF5, LiMnF4, Na3MnF6. Интересно, что ян-теллеровское искажение проявляется лишь у части комплексов (в NaK2MnF6 реализуются две длинные связи Mn F 2х0,206 и четыре короткие связи 4х0,186 нм; в [Cr(NH3)6]MnF6 все расстояния Mn-F одинаковы и равны 0,1922 нм).
Желтый Na2MnF6 образуется при электрохимическом окислении MnF2 в 40 %-=ой HF в присутствии NaF. Желтый K2MnF6 выделен диспропорционированием растворов К2MnО4 в 40 %-ой НF или при обработке K2MnО4 и KМnО4 фтором при 350 оС. Аналогичное соединение иона NO , (NO)2MnF6, получено обработкой трифторида MnF3 в жидком BrF3 солью межгалоидного соединения NOBrF4. Кроме комплексов состава М2MnF6, известны комплексы МMnF5 со щелочными металлами. Так, обработка свободным фтором KMnF3 при 500 оС дает KMnF5. Он же образуется при реакции перманганата KMnО4 с BrF3, причем количественно выделяется О2. Магнитные моменты этих соединений соответствуют чисто спиновым значениям. Двухвалентные катионы М2 (М = Mg, Ca, Sr, Ba, Zn, Cd, Hg) образуют комплексные соединения только одного типа MIIMnF6.
Фторид со смешанными валентностями Mn2F5 получен при испарении смеси MnF2 и MnF3 или их нагреванием в заваренной золотой ампуле и изучен недостаточно.
Тетрафторид марганца MnF4, образуется непосредственно из простых веществ при 600-700 оС. Чистый препарат имеет цвет от синего до сине-серого. MnF4 характеризуется очень высокой химической активностью, легко разлагается следами влаги. При комнатной температуре медленно диссоциирует на трифторид MnF3 и свободный фтор. Аналогично разлагается в вакууме уже при 0 оС.
С фторидами металлов он образует комплексные фтороманганаты(IV) – KMnF5 и K2MnF6. Их получают взаимодействием фторида MF с MnF2 в токе фтора, фторированием фтороманганатов(III), Na2MnF6 и (NH4)2MnF6 –электролитическим окислением дифторида MnF2 в 40 %-ом растворе HF. При обработке фтором растворов К2MnО4 или КMnО4 (350-375 оС) выпадает желтый осадок К2MnF6. В случае проведения диспропорционирования оксоманганата(VI) К2MnО4 в горячем растворе 40-% при охлаждении выделяется осадок К2MnF6:
3K2MnO4 8HF = 2KMnO4 K2MnF6↓ 2KF 4H2O.
Cоединение разлагается в водных растворах щелочей и карбонатов щелочных металлов с осаждением оксогидрата MnО(ОН)2. Из интенсивно окрашенного раствора К2MnF6 в соляной кислоте при нагревании выделяется хлор, щавелевая кислота мгновенно окисляется до СО2.
Другой фтороманганат(IV) КMnF5 получают из КMnF3 фторированием при 500 оС или интенсивно протекающим взаимодействием перманганата с BrF3. При этом количественно выделяется кислород:
3KMnO4 5BrF3 = 3KMnF5 6O2↑ 2,5Br2↑.
Выделены фтороманганаты(IV) щелочноземельных металлов и элементов 2 Группы состава МIIMnF6. Очень интересны соединения тетрафторида марганца с фторидами инертных газов. С ХеF2 получены комплексы XeMnF6 (XeF2.MnF4) и XeMn2F10 (XeF2.2MnF4). Соединения с гексафторидом ксенона более разнообразны: nXeF6.MnF4 (n = 4, 2, 1 и 0,5): Xe4MnF28, Xe2MnF16, XeMnF10 и XeMn2F14.
У марганца известен один оксофторид MnO3F. Формально его можно представить как продукт замещения в ионе MnO4— аниона кислорода на F—. MnO3F изоэлектронен с MnO4— (58 электронов, из них 24 валентных) и представляет собой искаженный тетраэдр (межатомные расстояния Mn-O 0,1586 нм и Mn-F 0,1724 нм, угол О-Mn-F 108,5 о). MnO3F получают взаимодействием перманганата с фторсульфоновой кислотой или с пентафторидом иода:
KMnO4 2HSO3F = MnO3F KSO3F H2SO4,
KMnO4 IF5 = MnO3F IOF3 KF.
Образующийся MnO3F конденсируют при – 80 оС. При 0 оС разлагается, часто с взрывом, на MnF2, MnO2 и О2. Во влажном воздухе интенсивно гидролизуется с выделением фиолетовых паров Mn2O7 или НМnO4, с избытком воды – до НMnO4 и НF. Довольно хорошо растворим в безводном НF. Обладает сильными окислительными свойствами, и используется в органической химии.
Галогениды технеция и рения практически не находят аналогий в химии марганца, так как многие из них представляют собой кластеры. Кратность связи металл-металл возрастает при понижении степени окисления (табл.5.6).
Таблица 5.6. Хлориды рения
| Формула | Цвет | Т. пл., °C | r(Re–Re), нм | Кратность связи Re–Re | Метод синтеза | Продукты взаимодействия с водой |
| Re3Cl9 | Темно-красный | 500 возг | 0,249 | 3ReCl5 = Re3Cl9 3Cl2 3ReCl5 3SnCl2= Re3Cl9 3SnCl4 3ReCl5 3H2= Re3Cl9 6HCl | Re3Cl9.2Н2О, Re2O3×xH2O | |
| β-ReCl4 | Черный | 300 разл | 0,273 и 0,437 | Re3Cl9 3ReCl5 = 6ReCl4* SbCl3 2ReCl5 = 2ReCl4 SbCl5 CCl2=CCl2 2ReCl5 ® 2ReCl4 C2Cl6 | ReO2×xH2O | |
| Re2Cl10 | Красно-коричн. | 0,373 | 400°C 2Re 5Cl2 = Re2Cl10 | HReO4 ReO2×xH2O | ||
| ReCl6 | Темно-зеленый | – | 500°C Re 3Cl2 = ReCl6 | HReO4 ReO2×xH2O |
* α-модификацию получают ReO2.2H2O 4SOCl2 = α-ReCl4 4SO2↑ 4HCl↑.
Среди галогенидов технеция и рения лучше всего изучены хлориды. Низший из известных хлоридов рения представляет собой треугольный кластер Re3Cl9. Атомы рения связаны друг с другом в треугольник двойными связями металл- металл и мостиковыми атомами хлора,которые лежат в плоскости треугольника (рис.5.43. Безводный хлорид рения(III) и продукт его гидратации). К каждому атому рения дополнительно присоединены по два атома Cl, связывающие отдельные кластеры в единый каркас. Вещество диамагнитно, что объясняется образованием двойных связей металл-металл (σ π) – расстояние Re-Re в кластере оказывается более коротким, чем в простом веществе (0,275 нм).
На воздухе хлорид рения(III) постепенно гидратируется, переходя в дигидрат Re3Cl9.2Н2О, в котором сохраняется кластерное ядро Re3Cl9, но отдельные треугольные кластеры изолированы друг от друга, благодаря координации молекул воды (рис.5.43). Водный раствор имеет красный цвет, свойственный молекулам Re3Cl9, но постепенно гидролизуется с выделением осадка гидратированного оксида Re2О3. В солянокислой среде гидролиз не происходит вследствие образования треугольных кластеров [Re3Cl12]3– (рис.5.43). Они могут бытьполучены в виде твердых комплексных солей.
При сплавлении с хлоридами металлов происходит диспропорционирование:
4[ReCl3]3 18KCl = 9K2ReCl6 3Re.
Бромид и иодид рения(III) также представляют собой трехъядерные кластеры.
ДОПОЛНЕНИЕ. Диядерные кластерные галогенидные комплексы технеция и рения.
При взаимодействии пертехнетатов и перренатов с фосфорноватистой кислотой в солянокислом растворе, либо при восстановлении хлоридных комплексов [МCl6]2– водородом в момент выделения образуются синие растворы, содержащие диядерные кластеры [М2X8]2–. Введением в раствор крупных катионов удается выделить кристаллические соли – сине-черные у технеция, голубые и зеленые в случае рения:
2KReO4 4H3PO2 8HBr 2CsBr = Cs2[Re2X8].2H2O 4H3PO3 2KBr 2H2O.
В комплексном ионе [M2X8]2– (Рис.5.44. Строение [Re2Cl8]2– (а) и cхема превращений октахлородиренатов(б)), восемь атомов галогена находятся в заслоненной конфигурации, располагаясь по вершинам тетрагональной призмы. Геометрия координационного окружения и элеткронное строение этих частиц напоминает описанные ранее карбоксилаты хрома(II) Cr2(RCOO)4(H2O)2 и рения(III) Re2(RCOO)4Cl2. Восемь валентных электронов двух атомов металла заселяют четыре связывающие МО, что соответствует четверной связи (1σ 2π 1δ) металл-металл. Эти связи отличаются повышенной прочностью и сохраняются при обратимых переходах из кристалла в раствор, при замещении экваториальных или аксиальных лигандов. Ионы [ReCl8]2– поддаются восстановлению и участвуют в разнообразных реакциях замещения лигандов (Рис. 5.44. б).
В отличие от рения, технеций образует устойчивые комплексы со смешанными степенями окисления, например, [Tc2Cl8]3–, Они выделяются при обработке растворов пертехнетата в конц. соляной кислоте около 100 оС металлическим цинком, или восстановлением таких же растворов водородом в автоклаве при 150 оС. В них технеций имеет степень окисления 2,5. Получены также двуядерные соединения K8[Tc2Cl8]3.4H2O и MI8[Tc2Cl8]3.2H2O (MI = NH4 , Cs ) со степенью окисления технеция 2,67.
(Ф.А. Коттон, Р. Уолтон, Кратные связи металл-металл, М., Мир, 1985)
КОНЕЦ ДОПОЛНЕНИЯ
Твердый тетрахлорид ReCl4 построен из октаэдров [ReCl6], попарно сочлененных общими ребрами в димеры со связью Re-Re, которые, в свою очередь, посредством мостиковых атомов хлора формируют бесконечные цепи (Рис.5.45. Строение тетрахлорида (а) и пентахлорида (б) рения). Тетрахлорид технеция также полимерен. Оба вещества представляют собой гигроскопичные кристаллы, разлагающиеся на воздухе и в щелочных растворах. В концентрированной НCl они образуют интенсивно окрашенные растворы, содержащие хлоридные комплексы.
Темно-красный кристаллический тетрахлорид ТсCl4 является наиболее устойчивым хлоридом технеция. Его синтез осуществляют прямым хлорированием, действием газообразного НCl на технеций при 350-500 оС или взаимодействием высшего оксида с парами СCl4. В отличие от тетрахлорида рения, в структуре TcCl4 расстояния между атомами технеция (0,362 нм) слишком велики для образования связей металл-металл. На отсутствие таких связей указывает также высокая магнитная восприимчивость соединения технеция. Тетрабромиды и иодиды рения получают восстановлением перренатов в водном растворе соответствующей галогеноводородной кислоты. Структура этих веществ неизвестна. Существование TcBr4 находится под вопросом, о TcI4 сведения отсутствуют.
Действием на пертехнетаты и перренаты сильным восстановителем в концентрированной соляной кислоте могут быть получены комплексные хлориды:
KTcO4 3KI 8HCl = K2TcCl6 3/2I2 2KCl 4H2O.
Реакция восстановления протекает по сложному механизму. Первичный продукт восстановления представляет собой хлоридный комплекс технеция(V), который диспропорционирует на TcO4– и [TcCl6]2–.
Гексахлоротехнециевая(IV) кислота выделена из раствора в виде кристаллогидрата H2[TcCl6].9H2O В ее структуре ионы [TcCl6]2–, имеющие форму слегка искаженного октаэдра, молекулы воды и ионы Н3О соединены водородными связями в зигзагообразные цепочки. Сильные гексахлоротехнециевая(IV) и рениевая(IV) кислоты образует соли со многими металлами. Гексахлоротехнетаты и -ренаты щелочных металлов и аммония растворимы в воде, с ростом размера катиона растворимость уменьшается. В растворах они медленно гидролизуются до диоксидов МO2.nH2O, проходя через промежуточные стадии [МCl5(H2O)]—, [МCl4(H2O)2] и [МCl3(H2O)3] .
Черно-коричневый хлорид рения(V) получают взаимодействием простых веществ. В твердом состоянии он состоит из димеров Cl4Re(μ-Cl)2ReCl4, в которых два октаэдра [ReCl6] соединены двумя мостиковыми атомами хлора (рис.5.45. б). При 150 оС начинается возгонка пентахлорида, сопровождающаяся его разложением на Re3Cl9 и хлор. В воде он диспропорционирует с осаждением гидратированного диоксида:
3ReCl5 12Н2О = НReО4 2ReО2.2Н2О l5НCl,
а в соляной кислоте образует зеленые растворы, содержащие комплексные хлориды:
3ReCl5 4Н2О = НReО4 2Н2ReCl6 3НCl.
При сплавлении с хлоридом калия образуется устойчивый гексахлороренат(IV):
2ReCl5 4КCl = 2К2ReCl6 Cl2↑.
Гексахлориды технеция и рения МCl6 синтезируют из простых веществ при повышенном давлении хлора. Подобно другим ковалентнрым галогенидам, на влажном воздухе они дымят, выделяя пары НCl. Гидролиз сопровождается диспропорционированием.
При нагревании на воздухе галогениды технеция и рения окисляются, превращаясь в оксохлориды:
6[ReCl3]3 21О2 = 6ReОCl4 12ReО3Cl 9Cl2↑,
4ReCl5 4О2 = 2ReОCl4 2ReО3Cl 5Cl2↑.
В целом оксохлориды технеция и рения более многочисленны, чем оксохлориды марганца, и более устойчивы. Их гидролиз сопровождается диспропорционированием.
3ReОCl4 9Н2О = 2НReО4 ReО2.2Н2О↓ 12НCl.
Чистый коричневый ReОCl4 получен взаимодействием сухого кислорода с пентахлоридом рения. Вещество состоит из молекул, координация атомов рения квадратно-пирамидальная с атомом кислорода в вершине. При нагревании до кипения оксохлорид разлагается. При облучении переходит в неустойчивый оксохлорид рения (V) ReОCl3.
Оксохлорид рения(VII) ReО3Cl – низкоплавкое легко летучее вещество (т. пл. 4,5 и т. кип. 130 оС), образующееся при действии хлороводорода на нагретый рениевый ангидрид, при хлорировании триоксида, окислении сульфида ReS2 смесью хлора и кислорода:
2ReS2 3Cl2 3O2 = 2ReO3Cl 2S2Cl2.
Во влажном воздухе дымит, подвергается быстрому гидролизу. Вступает во взаимодействие со многими металлами, в том числе, с ртутью и серебром. Обладает фотохимической активностью – на солнечном свету приобретает красно-фиолетовую окраску.
Коричневый TcОCl3 образуется при хлорировании диоксида ТсО2 или при фотохимическом распаде TcОCl4. При нагревании выше 500 оС сублимирует. Он легко гидролизуется и диспропорционирует по схеме 3TcV= 2TcIV TcVII. Красный, легко плавкий (т. пл.~35 оС) TcОCl4 получается при хлорировании металла в присутствии следов кислорода и отделяется от сопутствующих TcCl4 и TcОCl3 ступенчатой ваккуумной отгонкой в темноте. При комнатной температуре устойчив, при освещении быстро разрушается даже при – 78 оС. Разбавленным раствором аммиака гидролизуется до ТсО2 и ТсО4— в отношении 1:2. Бесцветный TcО3Cl синтезирован действием кислорода на TcCl4 и также экстракцией из ТсО4—, растворенного в конц. H2SO4. в присутствии ионов Cl—. Легко кипящая жидкость (т. кип. 25 оС). Подобно галогенидам, оксогалогениды образуют анионные комплексы. Некоторые из них используются как исходные вещества для синтеза по реакциям замещения лигандов:
[MOCl4]— 5NCS— = [MO(NCS)5]2- 4Cl—.
Группировка МО3 , содержащаяся в ионе [MOCl5]2-, напоминает ион ванадила VO2 . Она возникает при восстановлении ионов пертехнетатов и перренатов в холодной концентрированной соляной кислоте. В зеленых кристаллах (NH4)2[TcOCl5] длина апикальной двойной связи Tc═O составляет 0,172 нм. В воде соль мгновенно гидролизуется на TcO4— и [TcCl6]2-, а раствор HI быстро восстанавливает ее до производных TcIV.
Фторидные производные технеция включают два бинарных фторида ТсF5 и ТсF6. Пентафторид ТсF5 получают нагреванием (350 оС) тонкого порошка Тс в струе фтора, разбавленного азотом, или действием иода на раствор ТсF6.в пентафториде иода:
2TcF6 I2 = 2TcF5 2IF.
Полученный продукт, желтое твердое вещество, плавится при 50 оС, переходя в вязкую жидкость. При 60 оС начинает распадаться, образуя стекло. Изоструктурен с пентафторидами CrF5 и VF5: октаэдры TcF6 соединены в бесконечные цепи цис-фторидными мостиками. При взаимодействии хлоридов щелочных металлов MCl с TcF6 в растворе IF5 образует соли аниона [TcF6]— желтого цвета. Водой они разлагаются на [TcF6]2-, [TcO4]— и TcO2.
Гексафторид ТсF6 получают из простых веществ при 400 оС в никелевой аппаратуре, хранят в никеле или в стекле пирекс. При этом необходим избыток фтора. Представляет собой легкоплавкое твердое вещество золотистого цвета, растворимое в HF. При действии щелочи выделяет черный осадок диоксида:
3ТсF6 20NaOH = 2NaTcO4 TcO2↓ 18NaF 10H2O.
Несмотря на отсутствие бинарного тетрафторида ТсF4, получены соли иона фторотехнетата(IV). Сплавление галогенотехнетатов(IV), K2[TcCl6] и K2[TcBr6], c KHF2 или KH2F3 приводит к соли K2[TcF6], служащей предшественником для других соединений. Так, соли Na и NH4 получают из нее ионным обменом, а соли Rb , Cs и Ba2 — осаждением из водных растворов K2[TcF6], благодаря их меньшей растворимости. Ион [ТсF6]2- устойчив к гидролизу, разрушается только щелочными растворами или сплавлением со щелочами.
Бинарные фториды рения наиболее многочисленны, хотя низшие фториды, нелетучие ReF2 и ReF3, изучены совершенно недостаточно в силу их нестабильности. Тетрафторид ReF4 получают сопропорционированием — действием паров гексафторида на металлический Re при 550 оС. Помимо этого, образуется наряду с пентафторидом ReF5 при восстановлении гексафторида ReF6 летучими карбонилами металлов, например, W(CO)6. Пентафторид ReF5 может быть получен прямым взаимодействием простых веществ и восстановлением гексафторида ReF6 на вольфрамовой проволоке при 600 оС:
6ReF6 W = 6ReF5 WF6.
В твердом состоянии молекулы ReF5 димеризованы. Пентафторид — низкоплавкое летучее твердое вещество, легко гидролизующееся водой с одновременным диспропорционированием. С кислородом воздуха образует оксофторид ReОF3.
Гексафторид ReF6 синтизируют из простых веществ при 120 оС. Согласно спектроскопическим данным, атом рения здесь октаэдрически окружен шестью атомами фтора. Это наиболее легкоплавкий и летучий из фторидов рения. Соединение термически устойчиво, возгоняется без разложения. Энергично гидролизуется водой – гидролиз сопровождается диспропорционированием, с кислородом воздуха дает оксофториды ReО2F2 и ReОF4. Активно реагирует с оксидами, например, интенсивно разъедает кварц и стекло при 300 оС:
3ReF6 3SiO2 = 2ReО3F ReF4 3SiF4.
Светло-желтый гептафторид ReF7, высший фторид среди всех переходных металлов, получают также из простых веществ при повышенном давлении и температуре 400 оС. Спектроскопически установлено, что его молекула имеет форму пентагональной бипирамиды. Это легко летучее твердое вещество, интенсивно гидролизуется водой, химически активно. С кислородом воздуха образует ряд оксофторидов ReО3F, ReО2F3 и ReОF5.
У технеция известны два оксофторида, ТсOF4 и ТсO3F. Первый из них образуется наряду с фторидом ТсF5 при синтезе из простых веществ в случае загрязнения фтора примесями кислорода. Известны две его модификации – синяя и более летучая зеленая. Синий ТсOF4, изоструктурный ReOF4, построен искаженными октаэдрами TcO6, соединенными цис-мостиковыми атомами F. Зеленый ТсOF4 образован дискретными тримерами с треугольниками из атомов Тс, асимметрично связанных цис-мостиковыми атомами фтора (Тс-О 0,166 нм, Тс-F(концевой) 0,181 и Тс-F(мостиковый) 0,226 нм). Оба оксофторида чувствительны к влаге и легко гидролизуются водой. Желтый оксофторид ТсO3F синтезируют пропусканием фтора над ТсО2 в никелевой аппаратуре при 150 оС или растворением пертехнетата в безводном НF:
NH4TcO4 2HF = TcO3F NH4F↓ H2O.
Это низкоплавкое (т. пл. 18,3 оС) твердое вещество, стабильное при комнатной температуре, его хранят в никеле, поскольку оно медленно разъедает кварц. Водой гидролизуется до НТсО4 и НF. Под давлением фтора при 400 оС превращается в ТсF6. Оксофторид ТсO3F обладает свойствами донора иона фтора и из раствора АsF5 выделяет твердый [TcO3] [AsF6]—.
Оксофториды рения известны только для высших степеней окисления – от ReV до ReVII. У ReV известен один оксофторид состава ReOF3, у ReVI – два, ReO2F2 и ReOF4, и у ReVII получены три таких производных, ReO3F, ReO2F3 и ReOF5. Все они образуются при контакте бинарных фторидов рения с кислородом воздуха, либо при недостаточном удалении влаги и воздуха из сферы реакции и в некоторых случаях – даже при контакте фторидов со стеклом. Оксофториды — чрезвычайно гигроскопичные вещества, дымят во влажном воздухе (кроме ReОF3) и полностью гидролизуются водой с образованием смесей НReО4 и НF или с одновременным диспропорционированием. При растворении оксофторида ReОF3 в воде образуется синий раствор, содержащий комплексные оксофторо-ионы.
Комплексные соединения рения МI2ReF8 и МIReF7 получают действием жидкого гексафторида ReF6 на фториды щелочных металлов. На воздухе они гидролизуются и разлагаются. Розовый К2ReF8, синтезированный в инертном тефлоне, имеет квадратно-призматическую структуру. При гидролизе превращается в К[ReOF5].
Известны три бинарных бромида рения: Re3Br9, ReBr4 и ReBr5. Тримерный красно-коричневый Re3Br9 образуется при термическом распаде пентабромида ReBr5. Трибромид по структуре и свойствам похож на трихлорид, отличается лишь меньшей устойчивостью к гидролизу. Темно-красный тетрабромид ReBr4 синтезируют осторожным выпариванием раствора НReО4 в избытке кислоты НBr. Он сильно гигроскопичен и хорошо растворяется в воде, ацетоне и диэтиловом эфире. Темно-коричневый пентабромид ReBr5 получают прямым бромированием металлического рения при 650 оС. Термически нестоек, легко гидролизуется с диспропорционированием.
Оксобромиды рения, ReОBr3, ReОBr4 и ReО3Br менее устойчивы по сравнению с хлоридными аналогами. Их синтезируют реакциями оксидов рения с бромом или бромидов рения с кислородом. Оксобромид ReОBr3, имеет структуру, аналогичную оксохлориду технеция TcOCl3: цис-октаэдрическая цепочечная структура, в которой октаэдры ReО4Br2 соединены ребрами в бесконечные цепочки. Оксобромид ReO3Br представляет собой низкоплавкое летучее вещество (т. пл. 39,8 и т. кип. 155,3 оС). Все оксобромиды легко гидролизуются водой, при этом оксобромиды ReОBr3 и ReОBr4 диспропорционируют.
До настоящего времени выделены два бинарных иодида рения, тримерный трииодид Re3I9 и тетраиодид ReI4. Блестящие черные кристаллы Re3I9 получены термическим распадом тетраиодида или введением метилового спирта в смесь НReО4 и НI. Хотя трииодид также тримерный, как его хлоридный и бромидный аналоги, он менее термически стабилен. Трииодид слабо растворим в воде и разбавленных растворах кислот. При нагревании диссоциирует – при 170 оС до дииодида ReI2 и при 380 оС до моноиодида ReI, которые практически не изучены.
Черный тетраиоид ReI4 получен медленным выпариванием раствора НReО4 в избытке кислоты НI. Термически нестойкий, при нагревании диссоциирует. Сильно гигроскопичное вещество, хорошо растворимое в воде, в ацетоне и диэтиловом эфире.
Бинарные бромиды и иодиды технеция неизвестны, хотя могут быть стабилизированы в форме анионных комлпексов, например, K2[TcBr6], Cs2[TcI6]. При нагревании они распадаются на технеций, бромид щелочного металла и свободный бром. При разложении аммонийной соли выделяется свободный азот:
(NH4)2[TcBr6] = Tc Br2↑ 2NH4Br N2↑.
Свойства галогенидных комплексов рения изменяются от фторидных к иодидным – их термическая устойчивость сильно ослабевает. Так, К2[ReCl6] возгоняется без разложения около 1000 оС, тогда как K2[ReI6] уже при 300 оС разлагается.
Различно отношение Re(IV) и Tc(IV) по отношению к иону CN-. Cоль K2[TcI6] в метаноле при реакции с KCN образует темно-зеленый раствор, из которого осаждается красно-коричневый осадок K2[Tc(CN)6]. В аналогичных условиях соль K2[ReI6] диспропорционирует, выделяя смесь K4[ReIII(CN)7].2H2O и K3[ReVO2(CN)4].
Единственный оксобромид технеция TcОBr3 в виде серо-черных кристаллов получен обработкой диоксида ТсО2 или пертехнетата NH4TcO4 бромом при 350-400 оС. В кристаллическом состоянии образует цис-октаэдрическую полимерную цепочечную структуру типа NbOCl3. Соединение легко гидролизуется с диспропорционированием.