
Ацетон (диметилпропанон)
Школьной программой не предусматривается изучение кетонов. Однако некоторые учителя знакомят учащихся на уроках или в процессе внеклассной работы с окислением вторичных спиртов и практически наиболее важным представителем класса кетонов — ацетоном. Ниже приводятся опыты, которые могут быть использованы с этой целью.
Горение ацетона. Несколько капель ацетона наливают на крышку фарфорового тигля и поджигают. Ацетон горит слабо светящимся пламенем.
Растворимость ацетона в воде. К 2 мл воды добавляют равный объем ацетона. По встряхивании не удается установить расслоения жидкостей. Ацетон растворяется в воде.
Ацетон как растворитель смол и пластмасс. На этом свойстве ацетона основано его применение для изготовления лаков и для склеивания изделий из пластмасс.
а) В нескольких миллилитрах ацетона в пробирке растворяют при помешивании стеклянной палочкой столько целлулоида, чтобы раствор стал слегка вязким. Смачивают полученным лаком вату и натирают ею гладкий кусочек дерева — после улетучивания растворителя предмет оказывается «лакированным».
б) Два кусочка очищенной кинопленки или органического стекла (плексигласа) смачивают с концов ацетоном.
Через 1—2 мин накладывают концы пленки друг на друга и слегка сдавливают. После высыхания кусочки оказывают прочно склеенными ацетоном.
Отношение к аммиачному раствору оксида серебра. В две пробирки наливают одинаковые количества аммиачного раствора оксида серебра. В одну пробирку прибавляют раствор альдегида, в другую — такой же объем ацетона. Пробирки помещают одновременно в стакан с горячей водой. В пробирке с альдегидом появляется зеркало в пробирке с ацетоном восстановления серебра и, следовательно, окисления кетона не происходит.
Окисление ацетона. Ацетон не может быть окислен окисью серебра (см. предыдущий опыт), но он может быть окислен более энергичными окислителями.
Около 1 мл ацетона разбавляют в пробирке водой, приливают
серной кислоты, подогревают и вносят небольшими порциями измельченный перманганат калия, пока не перестанет исчезать его фиолетовая окраска. При нагревании раствора можно обнаружить по запаху пары уксусной кислоты.
При окислении происходит разрыв углеродной цепи и образование двух кислот — уксусной и муравьиной:
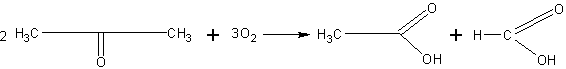
Получение бромацетона. Учащимся можно показать, что углеводородные радикалы кислородсодержащих органических веществ сохраняют в основном свойства углеводородов. Примером тому оказывается реакция бромирования ацетона:
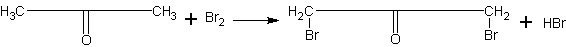
Реакция ацетона с бромом, кроме того, дает возможность довольно просто и в безопасной форме ознакомить учащихся с получением одного из слезоточивых веществ (лакриматоров). В настоящее время известны вещества, значительно превосходящие бромацетон по слезоточивому действию. Однако, принимая во внимание, что задача сводится к ознакомлению не с силой, а с характером действия вещества, целесообразно ограничиваться в указанных целях именно этим примером.
В вытяжном шкафу наливают в пробирку 1 мл ацетона и вносят несколько капель брома. Уже без подогрева обычно начинает ощущаться острый запах бромацетона. Если реакция не наступает, пробирку слегка подогревают (осторожно, беречь глаза!). Смачивают жидкостью из пробирки несколько полосок фильтровальной бумаги и раздают их на стеклышках учащимся или же помещают несколько капель жидкости на железную пластинку и нагревают ее над пламенем спиртовки.
Как только действие бромацетона на глаза обнаружено, хотя бы и в слабой форме, демонстрацию его прекращают и проветривают помещение.
Уравнение реакции учащиеся смогут составить сами, если им указать, что здесь в каждой молекуле ацетона замещается бромом один атом водорода.
В качестве наиболее сильного слезоточивого вещества указывается хлорацетофенон
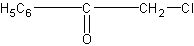
раздражающей концентрацией которого в воздухе считается 0,0003 мг/л.
Получение ацетона. В случае отсутствия в школьной лаборатории ацетон может быть получен для демонстрационных целей из солей уксусной кислоты, например, по реакции:
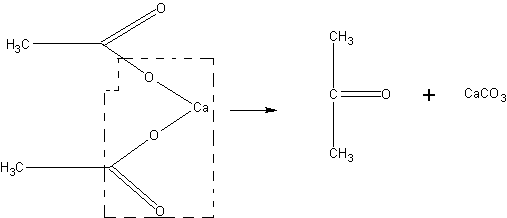
В наиболее простом виде опыт может быть проведен без выделения образующегося продукта.
В пробирке накаливают 2—3 г безводного ацетата натрия CH3COONa. Через 3—5 мин ощущается запах ацетона. Пары его при поджигании горят. По охлаждении пробирки с помощью соляной кислоты можно констатировать образование карбоната:
2CH3COONa à СН3 — СО — СН3 Na2CO3
Na2CO3 2НС! à 2NaCl Н2О СО2
ГЛАВА VII
КАРБОНОВЫЕКИСЛОТЫ
Изучение предельных одноосновных кислот лучше всего начинать со второго члена гомологического ряда — с уксусной кислоты. Эта кислота частично уже известна учащимся, она доступна для школы, на ней более характерно проявляется структура карбоновых кислот. Муравьиная кислота как первый член ряда имеет некоторые особенности в строении и свойствах, а поэтому менее удобна для первоначального ознакомления с классом кислот.
В ряду одноосновных кислот вслед за уксусной и муравьиной рекомендуется рассмотреть также высшие гомологи кислот. Это позволит создать у учащихся более полную картину о гомологическом ряде и не возвращаться к этим кислотам в последующей теме при изучении жиров.
Химические свойства кислот в настоящей главе приводятся не полностью. С целью избежать повторения реакции кислот со спиртами описываются в следующей главе, где они являются специальным предметом рассмотрения.
УКСУСНАЯ КИСЛОТА
Учащимся знаком запах уксусной кислоты, они имеют представление о некоторых химических свойствах ее, поскольку эти свойства являются общими со свойствами неорганических кислот, знают структурную формулу кислоты. Поэтому не особенно существенно, какой порядок ознакомления с веществом будет принят. Если учитель сочтет необходимым уделить внимание обоснованию структурной формулы кислоты, то после рассмотрения физических свойств можно перейти к химическим свойствам, так как именно они дадут ответ на интересующий вопрос, и лишь затем рассмотреть способы получения.
Напротив, если учитель хочет подчеркнуть генетическую связь между органическими веществами различных классов, он предпочтет сначала рассмотреть способы получения кислот, особенно путем окисления спиртов и альдегидов, а затем перейдет к изучению химических свойств.
Кристаллизация уксусной кислоты. В демонстрационном опыте трудно показать, что чистая уксусная кислота кристаллизуется при 16,7°С. Легче показать лишь сам факт «замерзания» ее при охлаждении. Наблюдение кристаллов объяснит учащимся, почему чистая уксусная кислота носит название ледяной.
1. В пробирку наливают несколько миллилитров уксусной кислоты (по возможности безводной) и помещают пробирку в ледяную воду. Вскоре при встряхивании наблюдают образование крупных блестящих кристаллов кислоты, напоминающих замерзшую воду. Если кислота не безводна, то она может не закристаллизоваться, в таком случае лучше пользоваться охладительной смесью снега с поваренной солью.
2. Вымораживанием кислоты из водного раствора легко получить чистую ледяную уксусную кислоту, которую можно хранить в качестве демонстрационного препарата. В небольшую колбочку наливают концентрированную уксусную кислоту и охлаждают в смеси снега с солью. Когда значительная часть жидкости закристаллизуется, выливают из колбы остаток жидкости — воду с некоторым количеством уксусной кислоты. Дают растаять замерзшей кислоте и снова помещают колбу в охладительную смесь. Когда часть жидкости снова закристаллизуется, опять сливают жидкость с кристаллов и повто-
ряют всю операцию еще раз. Так удается получить практически безводную уксусную кислоту, которую при понижении температуры очень легко переводить в кристаллическое состояние.
Горение уксусной кислоты. Проводя ряд аналогий между уксусной кислотой и минеральными кислотами, следует всякий раз подчеркивать особенность уксусной кислоты как кислоты органической, например ее горючесть.
Наливают в пробирку немного уксусной кислоты и нагревают в пламени горелки. При 118°С кислота кипит. Продолжая нагревание, поджигают пары лучинкой. Они горят слабо светящим пламенем. Учащиеся составляют уравнение реакции горения (в продуктах горения углекислый раз и вода) и дают объяснение слабой светимости пламени.
Отношение уксусной кислоты к окислителям. Легко показать, что, в отличие от спиртов и особенно альдегидов, уксусная кислота более устойчива по отношению к окислителям.
К разбавленному водному раствору чистой уксусной кислоты в химическом стакане прибавляют раствор перманганата калия и серной кислоты. Фиолетовая окраска раствора не исчезает.
Однако было бы неверным сделать заключение о неокисляемо-сти уксусной кислоты. Учащиеся уже знакомы с полным окислением кислоты при горении.
Действие уксусной кислоты на индикаторы. К растворам лакмуса и метилоранжа в демонстрационных пробирках или стаканчиках приливают немного раствора уксусной кислоты. Лакмус краснеет, метилоранж принимает розовую окраску (белый экран!).
Взаимодействие кислоты с металлами. В три пробирки наливают равные количества раствора уксусной кислоты. В одну пробирку помещают стружки магния, в другую — мелкие кусочки цинка, в третью — кусочки железной или медной проволоки. В первой пробирке наблюдают довольно энергичную реакцию. Убеждаются в том, что выделяющийся газ — водород, для чего пробирку закрывают пробкой с вертикальной трубочкой, собирают газ в перевернутую пробирку и поджигают его. Во второй пробирке реакция идет медленно и требует подогрев. Реакция с медью не идет даже при сильном нагревании.
Взаимодействие с основаниями. К очень разбавленному раствору щелочи, подкрашенному фенолфталеином, добавляют понемногу раствор уксусной кислоты, пока не обесцветится фенолфталеин. Составляют уравнение соответствующей реакции.
Аналогичный опыт может быть показан с гидроксидом кальция, взмученным в воде. При добавлении уксусной кислоты раствор становится прозрачным (ацетат кальция растворим в воде).
Взаимодействие с солями. Для опыта следует воспользоваться солью кислоты более слабой, чем уксусная, чтобы показать течение реакции до конца и образование соли уксусной кислоты. Наиболее удобны для этой цели карбонаты.
В маленькую колбу с отводной трубкой помещают 1—2 г поташа, соды или толченого мела и приливают 4—5 мл раствора уксусной кислоты. Колбочку закрывают пробкой и отводную трубку погружают в стакан с известковой водой. Известковая вода мутится от углекислого газа. Немного получившегося раствора можно выпарить на часовом стекле, чтобы наблюдать образование соли уксусной кислоты.
Уксусная кислота — кислота слабая. Чтобы показать учащимся, что уксусная кислота имеет небольшую степень диссоциации и является кислотой слабой, необходимо наряду с демонстрацией обычного опыта электропроводности ее поставить опыты, позволяющие провести хотя бы качественное сравнение скорости реакций уксусной кислоты и минеральных кислот.
а) В три пробирки помещают кусочки цинка и приливают равные количества соляной, серной и уксусной кислот (одинаковой концентрации). Замечают, что с уксусной кислотой реакция идет очень плохо или идет только при нагревании.
б) В три демонстрационные пробирки насыпают равные количества карбоната меди и прибавляют равные количества растворов соляной, серной и уксусной кислот одинаковой концентрации. Замечают, что в последнем случае скорость реакции наименьшая.
в) Если учащиеся знакомы с гидролизом солей, то можно показать гидролиз ацетата натрия и ацетата железа.
Последний опыт одновременно знакомит учащихся с характерной цветной реакцией уксусной кислоты.
К разбавленному раствору ацетата натрия приливают несколько капель фенолфталеина, и раствор разливают в две пробирки. Раствор в одной пробирке нагревают. Незначительное вначале порозовение фенолфталеина становится совершенно отчетливым при нагревании, так как гидролиз при этом усиливается:
CH3COONa Н2O ó СН3СООН NaOH
Поскольку водный раствор соли имеет щелочную реакцию, значит, уксусная кислота — кислота слабая.
Разбавленный раствор уксусной кислоты в стакане нейтрализуют постепенно щелочью в присутствии фенолфталеина или берут ранее полученный продукт нейтрализации. К раствору прибавляют несколько капель раствора хлорида железа FeCl3. Наблюдается характерное темно-красное окрашивание вследствие образования
ацетата железа Fe (III). При кипячении раствора образуется обильный красно-бурый осадок основного ацетата железа Fe(OH)2CH3COO. Гидролиз в этом случае проходит в значительной степени, так как соль образована слабым основанием и сравнительно слабой кислотой.
Основность уксусной кислоты. Хотя учащимся известны молекулярная и структурная формулы кислоты, можно экспериментально показать, что уксусная кислота одноосновна. Тогда станет совершенно очевидным различие в свойствах атомов водорода в молекуле кислоты.
Одноосновность кислоты легко показать, демонстрируя реакцию нейтрализации с применением молярных растворов.
Молярные растворы можно приготовить, например, из расчета 4 г едкого натра в 100 мл раствора и 6 г уксусной кислоты в 100 мл раствора. Отмеряют в коническую колбочку 10 мл молярного раствора кислоты, добавляют 2—3 капли фенолфталеина и прибавляют постепенно молярный раствор щелочи из другой бюретки до тех пор, пока появляющаяся розовая окраска не будет исчезать при взбалтывании жидкости, т.е. на каждую молекулу кислоты потребовалась одна, а не две и не три молекулы щелочи. Это значит, что уксусная кислота одноосновна.
Количественное получение метана из солей уксусной кислоты. Соли уксусной кислоты при нагревании со щелочью образуют метан (с. 27). Количественное проведение опыта дает возможность сделать заключение о структурной формуле кислоты.
Исследование продуктов реакции и сопоставление количества вступившей в реакцию соли с количеством образующегося метана позволяют сделают заключение о том, что реакция выражается уравнением:
CH3COONa NaOH à СН4 Na2CO3
На основании этого ведутся рассуждения о возможной структуре соли и, следовательно, кислоты.
Собирают прибор, как показано на рис. 30. В небольшую колбу или пробирку (желательно из тугоплавкого стекла) помещают хорошо перемешанную смесь порошков свежеприготовленного безводного ацетата натрия и натронной извести в весовом отношении 1 : 4 (например, 2 и 8 г или 3 и 12 г). Избыток извести необходим, чтобы обеспечить полное использование соли. Двугорлую склянку-газометр заполняют насыщенным раствором поваренной соли. Прибор проверяют на герметичность.
Смесь нагревают на газовой горелке. Нагревание должно быть равномерным и достаточно интенсивным. После того как объем
метана перестанет увеличиваться, нагревание прекращают и дают пробирке остыть. Некоторое количество раствора хлорида натрия при этом переходит обратно в склянку. Установив уровни жидкости в стакане и склянке на одной линии, определяют объем газа в склянке или объем жидкости в стакане.
Отделив газометр от пробирки, можно вытеснить из него часть газа (поднимая стакан с водой) и подтвердить, что этот газ — метан. Вычислив количество вещества исходной соли и количество вещества образующегося метана (н.у.), делают вывод, что из одного моля ацетата натрия получается один моль метана. Получающиеся отклонения из-за неполноты протекания реакции и образования побочных продуктов незначительны и совершенно не дают повода сделать какое-либо другое заключение (например, о том, что из одного моля соли образуется два моля метана).
Чтобы установить природу твердого продукта реакции, в пробирку добавляют немного соляной кислоты, закрывают пробкой и отводную трубку погружают в известковую воду. По помутнению известковой воды устанавливают, что при реакции образовался карбонат. Очевидно, что это карбонат натрия.
Рис. 30. Количественный опыт получения метана при взаимодействии ацетата натрия с натронной известью
Опыт дает надежные результаты в том случае, если исходные вещества будут тщательно подготовлены, хорошо перемешаны и будет обеспечено достаточное нагревание.
Получение кислоты окислением этанола. Альдегиды, как известно учащимся, легко окисляются в кислоты. Поэтому окисление спиртов в альдегиды можно поставить так, что образующийся альдегид, не успев удалиться из сферы реакции, тут же окислится далее в кислоту:
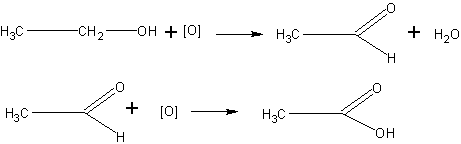
В качестве окислителя может быть использована хромовая смесь. Чтобы эта смесь могла окислить спирт в кислоту, реакцию проводят в условиях большей концентрации окислителя: берут больше бихромата, чем при получении альдегида, а спирт приливают к окислительной смеси постепенно, по мере отгонки образующейся кислоты.
Когда эти особенности получения кислоты обсуждены с учащимися, им становится понятным весь последующий опыт и одновременно закрепляются в памяти условия получения альдегидов из спиртов.
В колбочку с отводной трубкой (рис. 31) помещают 5 г измельченного бихромата калия К2Сг2О7 и 5 мл концентрированной серной кислоты. Из капельной воронки прибавляют по каплям водный раствор этанола (2 мл спирта на 8 мл воды) и отгоняют образующуюся уксусную кислоту. В приемнике собирается несколько миллилитров водного раствора уксусной кислоты с примесью альдегида и сложного эфира. Учащиеся должны дать объяснение изменению окраски раствора в колбе.

По запаху убеждаются в получении кислоты. Наблюдают образование водорода при действии на кислоту магния. Если учащиеся были знакомы также с цветной реакцией уксусной кислоты, то
нейтрализуют часть дистиллята содой (реакция не должна быть кислой) и добавляют несколько капель раствора хлорида железа (III). Темно-красное окрашивание раствора свидетельствует о наличии уксусной кислоты.
Запах уксусной кислоты иногда перебивается запахом альдегида. Чтобы освободиться от альдегида, кислоту нейтрализуют содой и раствор выпаривают. Альдегид при этом улетучивается. К остатку приливают минеральную кислоту и смесь слегка нагревают. Ощущается характерный запах уксусной кислоты.
Получение уксусной кислоты из ее солей. Данный эксперимент является повторением хорошо известного учащимся опыта из курса неорганической химии.
В колбочку с отводной трубкой насыпают 5 г обезвоженного ацетата натрия и добавляют 5 мл концентрированной серной кислоты. Сразу же начинается реакция. Подогревая колбу на горелке, отгоняют уксусную кислоту в пробирку через воздушный холодильник (стеклянную трубку). Если пробирку держать в ледяной воде, можно получить чистую уксусную кислоту в кристаллическом виде.
При необходимости уксусная кислота может быть получена таким способом’ в значительных количествах.
Получение кислоты из продуктов сухой перегонки дерева. Ранее было показано (с. 86), что сухая перегонка дерева приводит к выделению дегтярной воды. Поэтому задача получения уксусной кислоты сводится к тому, чтобы удалить из дегтярной воды метанол и ацетон.
Предварительно с помощью лакмуса необходимо убедиться в присутствии кислоты в дегтярной воде.
Имеющееся в распоряжении количество дегтярной воды обрабатывают взмученной в воде известью до появления щелочной реакции на лакмус. При этом уксусная кислота образует ацетат кальция (СН3СОО)2Са. От раствора отгоняют метанол и ацетон или же раствор выпаривают до образования сухого остатка — так называемого древесного порошка, представляющего собой ацетат кальция с избытком извести. Порошок обрабатывают в колбе концентрированной серной кислотой и отгоняют уксусную кислоту, как в предыдущем опыте. В приемнике собирается концентрированная (60—80-процентная) уксусная кислота.
Получение уксусного ангидрида. Учащиеся должны иметь представление о том, что карбоновым кислотам, как и минеральным кислородсодержащим кислотам, соответствуют ангидриды.
При отсутствии в школьной лаборатории уксусного ангидрида (СН3СО)2О для демонстрационных целей он может быть приготовлен по реакции ацетата натрия с хлористым ацетилом:
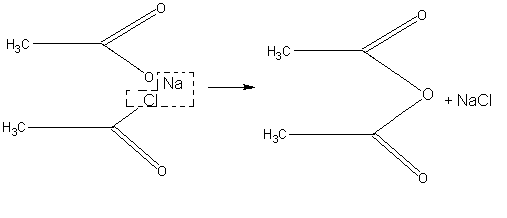
Чтобы жидкий хлористый ацетил по возможности нацело прореагировал и не улетучивался с ангидридом, пользуются избытком нелетучего вещества — ацетата натрия. Опыт проводят в вытяжном шкафу.
В колбу с отводной трубкой помещают 8 г обезвоженного ацетата натрия и приливают маленькими порциями 4—5 мл хлористого ацетила, встряхивая при этом колбу. Реакция образования ангидрида идет с выделением тепла.
К колбе присоединяют воздушный холодильник и отгоняют образующийся ангидрид на пламени горелки (темп. кип. 140°С). Сперва колбу нагревают осторожно, затем нагревание усиливают. Так как ангидрид обычно получается все же с примесью хлористого ацетила, к дистилляту прибавляют 0,5—1 г порошка ацетата натрия, чтобы полностью превратить хлористый ацетил в ангидрид, и снова перегоняют или же сохраняют его с примесью этой соли. Препарат обладает запахом уксусной кислоты. Хранят его в плотно закупоренной склянке. При действии воды на уксусный ангидрид образуется уксусная кислота:
(СН3СО)2O Н2О à 2СН3СООН
О наличии последней убедительно свидетельствует лакмусовая бумажка.
Получение хлористого ацетила. Хлористый ацетил может оказаться необходимым не только для получения ангидрида уксусной кислоты, но и для синтеза сложных эфиров.
Его получают действием треххлористого фосфора на уксусную
кислоту:
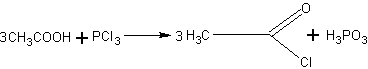
В колбу, соединенную с нисходящим водяным холодильником, помещают ледяную уксусную кислоту и при охлаждении небольшими порциями прибавляют треххлористый фосфор. Затем колбу закрывают пробкой и нагревают в вытяжном шкафу на водяной бане при 45— 50°С. Нагревание ведут до тех пор, пока не прекратится выделение хлороводорода (вследствие взаимодействия трех-
хлористого фосфора с протонсодержащими реагентами) и пока жидкость не разделится на два слоя. Сверху собирается хлористый ацетил, а внизу — фосфористая кислота. Хлористый ацетил отгоняют на водяной бане.
Так как хлористый ацетил чрезвычайно легко разлагается влагой воздуха, то приемник следует закрыть хлоркальциевой трубочкой.
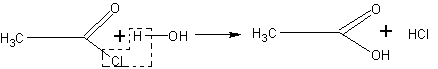
Хлористый ацетил можно отделить от фосфористой кислоты также с помощью делительной воронки.
Непременным условием успеха опыта является использование совершенно сухой посуды. Хранят хлористый ацетил в сухой склянке с плотной пробкой, залитой парафином.
Реакция хлористого ацетила с водой может служить наглядным примером взаимного влияния атомов в молекуле. Чрезвычайная подвижность атома хлора в группировке
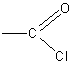
объясняется смещением электронной плотности в сторону кислорода карбонильной группы.
К 5—6 мл холодной воды в пробирке прибавляют несколько капель хлористого ацетила. Хлористый ацетил энергично реагирует с водой, образуя уксусную и соляную кислоты. В этом легко убедиться с помощью лакмуса и раствора нитрата серебра. Если вода для опыта взята недостаточно холодная, реакция происходит мгновенно. Разумеется, никакого подогрева смеси при этом не производится. Уксусный ангидрид реагирует с водой значительно слабее.
Исследование образца уксусной кислоты. Работа представляет интерес для внеклассных занятий по химии, так как знакомит учащихся с приемами контроля химической продукции.
Уксусная кислота может быть различной концентрации и может содержать примесь минеральных кислот, спирта и других органических веществ. Зная способ получения кислоты, учащиеся могут объяснить, как появляются эти примеси.
Концентрацию уксусной кислоты определяют титрованием ее щелочью известной концентрации. Для титрования столового уксуса может быть приготовлен 0,1 М раствор щелочи, для титрования эссенции — молярный раствор. С помощью бюретки берут по
5 мл исследуемого раствора в три конические колбочки, добавляют по 2—3 капли фенолфталеина и титруют из бюретки щелочью до появления остающейся при взбалтывании розовой окраски фенолфталеина. Из трех определений берут среднее арифметическое. Зная, что реакция выражается уравнением
СН3СООН NaOH à CH3COONa Н2О
вычисляют молярную, а затем и процентную концентрацию кислоты в исследуемом образце.
Примесь серной кислоты определяют, добавляя к образцу раствор хлорида бария. При отсутствии серной кислоты не должно получаться мути сульфата бария.
Примесь соляной кислоты устанавливают с помощью раствора нитрата серебра. В случае наличия хотя бы незначительного количества соляной кислоты при добавлении раствора нитрата серебра и подкислении азотной кислотой появляется муть или осадок.
Примесь органических соединений устанавливается с помощью слабого (0,1%) раствора перманганата калия. При наличии примесей перманганат обесцвечивается; уксусная кислота в этих условиях, как известно, не окисляется.
Таким способом могут быть исследованы образцы химически чистой уксусной кислоты, столового уксуса, уксусной эссенции и кислоты, полученной учащимися в лаборатории.
МУРАВЬИНАЯ КИСЛОТА
При изучении муравьиной кислоты необходимо поставить опыты, показывающие особенности ее строения и химических свойств.
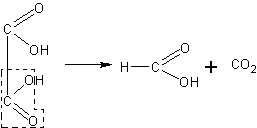
Разложение муравьиной кислоты на оксид углерода (II) и воду. После того как учащиеся ознакомятся с физическими свойствами муравьиной кислоты (агрегатное состояние, запах, цвет) и вспомнят ее структурную формулу по реакциям окисления формальдегида, целесообразно поставить вопрос о том, как может быть подтверждена ее структурная формула. Учащиеся обращают внимание на то, что молекула муравьиной кислоты построена как бы из молекулы воды и молекулы оксида углерода (II):

Естественно встает вопрос, нельзя ли получить кислоту реакцией оксида углерода (II) с водой. Учитель сообщает, что эта реак-
ция не идет так просто, как кажется: угарный газ не вступает в реакцию с водой. Но можно провести обратную реакцию: разложить кислоту на оксид углерода (II) и воду. В качестве водоотнимающего средства можно воспользоваться часто применяемой для этой цели серной кислотой. Оксид углерода (II) нетрудно будет обнаружить по его характерному пламени при горении.
В небольшую колбочку (или пробирку) с отводной трубкой наливают 1 мл муравьиной кислоты и 3-4 мл концентрированной серной кислоты. Колбу закрывают пробкой и нагревают в пламени горелки. Образующийся газ собирают в два цилиндра над водой. Во втором цилиндре газ поджигают (в первом — смесь газа с воздухом может дать взрыв). По синей окраске пламени учащиеся узнают оксид углерода (II). После этого поджигают оксид углерода (II) у отводной трубки. Опыт ведут до полного разложения муравьиной кислоты или останавливают его, прекращая нагревание. При проведении опыта необходимо следить, чтобы ядовитый газ не проникал в помещение. Опыт подтверждает формулу муравьиной кислоты.
Окисление муравьиной кислоты. Учащиеся легко устанавливают, что муравьиная кислота одновременно является альдегидом, так как имеет группировку атомов
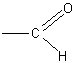
Это дает возможность предположить, что муравьиная кислота должна окисляться легче уксусной, и в частности давать реакцию серебряного зеркала.
До проведения опыта можно высказать предположение и о том, какие вещества будут получаться в результате реакции. Учащиеся составляют уравнение реакции, аналогичное уравнению для альдегидов, и замечают, что образуется угольная кислота:
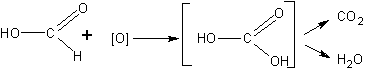
Поскольку угольная кислота, как известно, распадается с образованием углекислого газа, то для большей убедительности опыт, следовательно, надо поставить так, чтобы попытаться обнаружить углекислый газ.
а) Окисление перманганатом калия. В колбочке или пробирке с отводной трубкой к 1 мл муравьиной кислоты приливают 10 мл разбавленного раствора перманганата калия и немного серной кис-
лоты. Нагревают колбу через асбестовую сетку. Отводную трубку опускают в известковую воду. Перманганат калия при реакции обесцвечивается, содержимое колбы пенится от образующегося газа, и известковая вода мутится. Уксусная кислота, как известно, не обесцвечивает перманганат калия.
б). Окисление аммиачным раствором гидроксида серебра. Реакцию проводят как обычно, однако восстанавливающееся серебро при этом чаще всего выделяется не в виде зеркала, а в виде черного
осадка.
Получение муравьиной кислоты. Получение муравьиной кислоты из спирта и альдегида после всего изученного не представляет особого интереса. Значительно важнее было бы показать учащимся получение кислоты синтезом формиата натрия из оксида углерода(II) и едкого натра, что соответствует промышленному способу. Однако необходимость обращения со значительными количествами оксида углерода (II), даже при всей тщательности работы, делает опыт неприемлемым для школы.
При отсутствии в лаборатории муравьиной кислоты она может быть приготовлена нагреванием щавелевой кислоты с глицерином. Щавелевая кислота в этих условиях разлагается, образуя углекислый газ и муравьиную кислоту:
В колбу емкостью 200—250 мл с отводной трубкой помещают 30 г безводного глицерина и 30 г безводной щавелевой кислоты (кислоту обезвоживают осторожным нагреванием в сушильном шкафу при 95 — 100°С). Закрывают колбу пробкой с термометром, шарик которого погружают в смесь веществ. К колбе присоединяют нисходящий холодильник и приемник для дистиллята. Нагревают колбу через асбестированную сетку так, чтобы термометр показывал 110 — 115°С. Около 100°С начинается обильное выделение углекислого газа. При этом начинает отгоняться муравьиная кислота. Когда выделение углекислого газа станет слабым, добавляют вторую порцию (30 г) щавелевой кислоты и снова продолжают перегонку. При необходимости добавляют третью такую же порцию кислоты. После повторной дистилляции удается получить довольно крепкую муравьиную кислоту (темп. кип. кислоты 100°С).
Если почему-либо не удалось приготовить безводную щавелевую кислоту, можно воспользоваться кристаллической кислотой, беря ее всякий раз несколько большее количество. В этом случае будет получаться менее концентрированная кислота, так как одновременно перегоняется значительное количество воды.
Взаимодействие формиата натрия с натронной известью. Учащимся известно, что при нагревании солей карбоновых кислот с натронной известью (или едким натром) образуются соответствующие углеводороды.
Будет очень интересно предложить учащимся решить вопрос о том, какие продукты получатся при нагревании формиата натрия с натронной известью, а затем поставить соответствующий опыт и определить образующиеся вещества.
При реакции получаются водород и карбонат натрия:
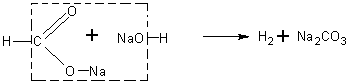
§
При изучении высших кислот учащиеся подробно знакомятся со свойствами бытового химического продукта — мыла и впервые на практике встречаются с веществом, обладающим двойственной функцией, — олеиновой (непредельной) кислотой.
Свойства стеариновой кислоты. Стеариновая кислота в воде нерастворима, поэтому для опытов, иллюстрирующих ее кислотные свойства, нельзя, как обычно, воспользоваться водным раствором.
а) Расплавляют немного стеарина или стеариновой свечи в фарфоровой чашке и помещают в него небольшой кусочек натрия. Наблюдается выделение газа — водорода:
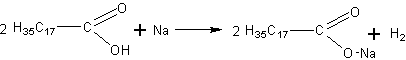
б) В пробирке растворяют кусочек стеарина в органическом растворителе (эфир, хлороформ, бензол) и к полученному раствору добавляют 2 капли спиртового раствора фенолфталеина. Затем вносят 1—2 капли разбавленного раствора гидроксида натрия и встряхивают пробирку. Розовой окраски раствора не наблюдается, так как стеариновая кислота нейтрализовала щелочь:
С17Н35СООН NaOH à C17H35COONa Н2О
При дальнейшем прибавлении щелочи к раствору кислоты розовая окраска появляется после того, как кислота будет нейтрализована.
Добавляя щелочь к эфиру с фенолфталеином без стеариновой кислоты, наблюдают моментальное появление окраски.
в) В пробирку со спиртом добавляют 2—3 капли щелочи и 1-2 капли фенолфталеина и к полученной смеси при перемешивании прибавляют эфирный раствор стеариновой кислоты или даже твердый стеарин. При этом происходит обесцвечивание раствора вследствие нейтрализации щелочи.
Стеариновая кислота — кислота слабая. О силе стеариновой кислоты можно судить по гидролизу ее солей, например стеарата натрия C17H35COONa.
а) Растворяют в воде тонкие стружки простого мыла и добавляют спиртовой раствор фенолфталеина. Появление розовой окраски говорит о том, что стеариновая кислота — кислота слабая. Параллельно может быть испытан фенолфталеином водный раствор ацетата натрия. В этом случае розовая окраска если и появляется, то очень слабая, так как уксусная кислота сильнее стеариновой.
б) В цилиндр до половины наливают воду, а затем по стенке осторожно прибавляют небольшое количество спиртового раствора мыла с фенолфталеином. В спиртовом растворе гидролиз соли не происходит и розовая окраска не наблюдается. На границе же соприкосновения спиртового раствора соли с водой появляется розовое кольцо.
Получение мыла (стеарата натрия) из стеарина. Один из современных способов получения мыла состоит в нейтрализации кислот, получаемых окислением синтетических углеводородов или же непосредственным синтезом из водяного газа.
Кусочек стеарина (стеариновой кислоты) нагревают в химическом стаканчике с водой, пока стеарин не расплавится (темп. пл. 69°С), и приливают к нему понемногу раствор гидроксида натрия до полного растворения (стеарат натрия растворяется в воде). К полученному раствору прибавляют поваренную соль до насыщения.
В насыщенном растворе поваренной соли стеарат натрия не растворяется и всплывает наверх в виде хлопьев.
Получение стеариновой кислоты из мыла. Стеариновая кислота, как кислота слабая, может быть получена действием минеральной кислоты на мыло:
2C17H35COONa H2SO4 à 2C17H35COOH Na2SO4
Как вещество не растворимое в воде, стеариновая кислота при этом выделяется из раствора.
В химическом стаканчике при нагревании готовят крепкий раствор мыла в воде. Для более быстрого растворения мыло должно быть предварительно нарезано в виде тонких стружек. Продолжая нагревать раствор мыла, прибавляют к нему раствор серной кислоты до выделения стеариновой кислоты. При охлаждении раствора 1 сверху образуется твердый слой стеариновой кислоты.
Таким способом в случае необходимости можно получить некоторое количество кислоты для классных занятий.
Моющее действие мыла. Мыло обладает свойством эмульгировать жиры и суспензировать твердые частички грязи.
а) Готовят, как было указано выше, раствор мыла в воде и прибавляют к нему 1 мл растительного масла. Смесь сильно взбалтывают до образования эмульсии, похожей на молоко. В другой пробирке подобным способом получают эмульсию из таких же количеств чистой воды и масла. Хотя во второй пробирке эмульсия была получена позднее, она быстро расслаивается на воду и масло. В первой же пробирке эмульсия оказывается устойчивой и не разделяется в течение всего урока.
б) В двух разных пробирках готовят суспензию сажи в воде и в мыльном растворе. Фильтруют растворы одновременно через бумажные фильтры. В первом случае фильтруется чистая вода, во втором случае мыльный раствор настолько сильно удерживает сажу, что суспензия, не разделяясь, проходит через фильтр.
Моющее действие мыла проявится еще более ярко, если фильтр с осадком сажи перевернуть на другую сторону, снова вложить в воронку и сперва пропустить через него воду, а затем мыльный раствор. Вода проходит прозрачной и не смывает сажу, мыльный же раствор совершенно очищает фильтр от сажи, становясь черным.
Действие жесткой воды на мыло. В жесткой воде мыло не дает пены и плохо мылит, так как превращается в нерастворимые в воде соли:
2C17H35COONa СаСI2 à (C17H35COO)2Ca 2NaCl
Хлопья нерастворимого мыла можно часто наблюдать при получении мыльного раствора в домашних условиях.
а) Раствор мыла в воде разливают в две пробирки. В одну из пробирок добавляют небольшое количество раствора хлорида кальция СаСI2. При встряхивании пробирок в одной из них наблюдают образование пены, в другой — отсутствие пены и образование осадка соли кальция.
б) В один цилиндр наливают около 50 мл дистиллированной воды, в другой такой же цилиндр — 50 мл водопроводной воды (если водопроводная вода очень «мягкая», в нее добавляют немного
раствора хлорида кальция). В оба цилиндра приливают по 1 мл спиртового раствора мыла и встряхивают. В первом случае образуется устойчивая пена. Во втором пены нет, но образуется осадок нерастворимой соли. Во второй цилиндр продолжают добавлять раствор мыла до тех пор, пока при встряхивании не будет получаться неисчезающая пена. По количеству затраченного раствора мыла можно приблизительно судить о степени жесткости воды.
Опыт показывает, что при пользовании жесткой водой большое количество мыла расходуется на осаждение кальциевых (и магниевых) солей и что только после их осаждения мыло, как поверхностно-активное вещество (ПАВ) начинает выполнять свою основную функцию — давать пену. Чтобы сделать воду «мягкой» и не расходовать мыло нерационально, соли кальция, как известно, осаждают предварительно кипячением или с помощью специальных добавок, например соды.
§
Из класса непредельных одноосновных карбоновых кислот наибольший интерес для изучения представляют акриловая и олеиновая кислоты. Первая — как первый член гомологического ряда, производные которой имеют большое значение при получении синтетических материалов. Олеиновая кислота является представителем непредельных кислот, входящих в состав жиров.
Вместо акриловой кислоты СН2=СН—СООН можно воспользоваться ее ближайшим и более доступным гомологом — метакриловой кислотой, эфиры которой применяются для получения широко известных полимерных продуктов (органического стекла).
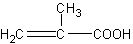
Получение метакриловой кислоты (по В.В.Некрасову). Метакриловая кислота может быть получена гидролизом сложного эфира — метилметакрилата. Метилметакрилат для этого опыта может быть в свою очередь получен деполимеризацией полиметилметакрилата (органического стекла), как это описано на с. 68. Гидролиз проводится в присутствии щелочи. Метакриловая кислота при этом образует соль, из которой она может быть выделена действием более сильной кислоты:
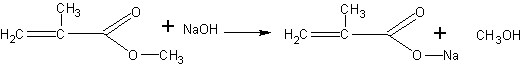
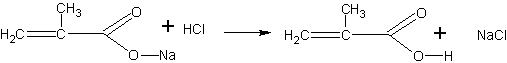
Помещают в пробирку 2 мл метилового эфира метакриловой кислоты (метилметакрилата) и 4 мл концентрированного раствора едкого натра. Чтобы смесь получилась гомогенной, добавляют 1 мл этанола и встряхивают. Через 15—20 мин переливают жидкость в фарфоровую чашку и выпаривают досуха на слабом пламени. Остаток представляет собой натриевую соль метакриловой кислоты с возможной примесью щелочи. Дальнейшая задача сводится к выделению кислоты из соли. Для этого сухой остаток переносят в пробирку и, охлаждая ее водой, добавляют несколько миллилитров концентрированной соляной кислоты (до кислой реакции на индикатор). После отстаивания смеси метакриловая кислота собирается сверху в виде маслянистой жидкости с характерным запахом. Кислоту переносят при помощи пипетки в другую пробирку.
Свойства метакриловой кислоты. Метакриловая кислота, будучи соединением непредельным, реагирует с бромной водой, перманганатом калия и легко полимеризуется.
а) К небольшому количеству (нескольким каплям) метакриловой кислоты приливают бромную воду и смесь встряхивают. Бромная вода обесцвечивается вследствие присоединения брома по месту двойной связи. Учащиеся составляют уравнение реакции.
Подобным образом проводится опыт с водным раствором пер-манганата калия.
б) К небольшому количеству метакриловой кислоты добавляют несколько крупинок перекиси бензоила в качестве инициатора реакции и нагревают до начала кипения. Кислота быстро полимеризуется.
Непредельность олеиновой кислоты. Олеиновая кислота является единственным представителем класса непредельных кислот, входящих в состав жиров и изучаемых в школьном курсе химии. Непредельность ее может быть проиллюстрирована действием обычных реактивов на двойную связь: бромной воды и раствора перманганата калия.
К 5—8 мл олеиновой кислоты в пробирке или колбочке приливают равный объем бромной воды или раствора перманганата калия и встряхивают. Окраска брома и перманганата калия исчезает вследствие присоединения по месту двойной связи.
ЩАВЕЛЕВАЯ КИСЛОТА
Наибольшее значение в этом разделе имеют опыты, связанные со строением щавелевой кислоты и ее двухосновностью.
Получение щавелевой кислоты из муравьиной. Первым подтверждением структурной формулы щавелевой кислоты может служить получение ее солей из солей муравьиной кислоты.
Формиат натрия при нагревании выделяет водород; одновременно образуется оксалат натрия
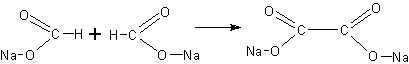
Очевидно, что из молекулы соли при этом уходит единственный атом водорода. А так как радикалы неспособны к сколько-нибудь длительному существованию, то они рекомбинируются, образуя молекулы новой соли, которой отвечает кислота
В пробирке, закрытой пробкой с вертикальной трубкой, нагревают в пламени горелки сухой формиат натрия.
Соль плавится и затем (при сильном нагревании) разлагается. Устанавливают выделение водорода, собирая его в пробирку или поджигая через некоторое время у отводной трубки.
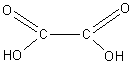
Разложение щавелевой кислоты при нагревании с серной кислотой. Если щавелевая кислота имеет формулу
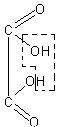
то можно предположить, что при отнятии от нее элементов воды должны выделяться углекислый газ и оксид углерода (II). Этот факт послужил бы подтверждением структурной формулы кислоты.
Учащимся можно предложить самим придумать прибор, в котором следовало бы провести эту реакцию так, чтобы обнаружить оба образующихся газа.
Опыт может быть осуществлен, например, в приборе, изображенном на рис. 32.
В небольшую колбочку помещают 2—3 г щавелевой кислоты и двойной объем концентрированной серной кислоты. В пробирку с отводной трубкой наливают известковую воду. При нагревании смеси кислот на горелке известковая вода мутится. Когда можно будет предположить, что воздух вытеснен из прибора, поджигают у отводной трубки пробирки оксид углерода (II), который горит характерным синим пламенем.
 Учащимся можно предложить разработать количественное проведение этого опыта, и в частности определить объемное соотношение образующихся газов. Некоторые указания к конструированию прибора им даст описание опыта разложения уксусной кислоты на углекислый газ и метан (с. 115). При этом в качестве обязательного требования необходимо поставить достижение максимальной простоты прибора.
Учащимся можно предложить разработать количественное проведение этого опыта, и в частности определить объемное соотношение образующихся газов. Некоторые указания к конструированию прибора им даст описание опыта разложения уксусной кислоты на углекислый газ и метан (с. 115). При этом в качестве обязательного требования необходимо поставить достижение максимальной простоты прибора.
Окисление щавелевой кислоты. Щавелевая кислота окисляется легче уксусной. Распадаясь на воду, углекислый газ и оксид углерода (II), она, очевидно, нуждается в незначительном количестве кислорода, чтобы дать конечные продукты окисления — углекислый газ и воду. Для простоты можно считать, что действие окислителя сводится к превращению угарного газа в углекислый.
В качестве окислителя может быть взят не раз применявшийся в подобных случаях перманганат калия с серной кислотой.
В небольшую колбочку с отводной трубкой наливают 5 мл концентрированного раствора щавелевой кислоты и равный объем разбавленной серной кислоты (1 : 5).
Из капельной воронки в колбу приливают постепенно 10 мл не слишком слабого раствора перманганата калия. При нагревании колбы фиолетовая окраска быстро исчезает и наблюдается энергичное выделение углекислого газа. Его пропускают в известковую или баритовую воду или же собирают в цилиндр и затем испытывают соответствующим способом. Уравнение реакции можно представить следующим образом:
5Н2С2O4 2KMnO4 3H2SO4 à 10СО2 K2SO4 2MnSO4 8Н2O
Образование кислых и средних солей щавелевой кислоты. В молекуле щавелевой кислоты имеются две карбоксильные группы, следовательно, щавелевая кислота двухосновная.
Как кислота двухосновная, щавелевая кислота может давать соли кислые и средние. Это может быть показано на реакции нейтрализации.
Растворяют 12 г щавелевой кислоты примерно в 20 мл воды и делят раствор пополам. К одной части раствора приливают понемногу концентрированный раствор гидроксида калия. Наблюдают, что сначала образуется осадок, который при дальнейшем прибавлении щелочи растворяется. Очевидно, что сначала получается кислая соль (гидрооксалат калия), нерастворимая в воде. В дальнейшем, при добавлении щелочи, эта соль превратилась в среднюю, растворимую в воде:
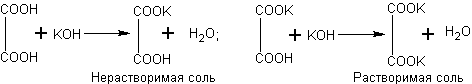
Если теперь к раствору средней соли прилить вторую порцию щавелевой кислоты, то снова образуется обильный осадок гидрооксалата калия:
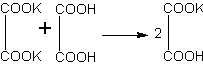
БЕНЗОЙНАЯ КИСЛОТА
Изучение бензойной кислоты на уроке не входит в программу по химии. Учителя иногда посвящают несколько минут ознакомлению с нею лишь для того, чтобы показать, как образуются кислоты в ароматическом ряду и что они собой представляют. Если ранее ставился опыт окисления бензальдегида в бензойную кислоту, здесь можно познакомить учащихся лишь с физическими свойствами ее.
Растворимость бензойной кислоты в воде. Встряхивая в пробирке очень небольшое количество кристаллов бензойной кислоты с водой, убеждаются, что заметного растворения не происходит. При нагревании пробирки происходит растворение кислоты, а при охлаждении она вновь кристаллизуется. Убеждаются, что водный раствор бензойной кислоты окрашивает лакмус в красный цвет.
Растворимость бензойной кислоты в щелочах. К небольшому количеству бензойной кислоты в пробирке прибавляют воду. Убедившись, что кислота не растворяется, добавляют по каплям крепкий раствор щелочи, пока кислота не растворится. После этого подкисляют раствор соляной кислотой. Выделяются мелкие кристаллы бензойной кислоты:
С6Н5СООН NaOH à C6H5COONa НСI H2O
C6H5COONa НСI à С6Н5СООН NaCl

Возгонка бензойной кислоты. Собирают прибор из двух пробирок разной величины, как показано на рис. 33. В большую пробирку на дно насыпают бензойную кислоту, а маленькая пробирка наполнена холодной водой. При нагревании прибора на пламени горелки бензойная кислота возгоняется и осаждается в виде белых кристаллов на пробирке-холодильнике.
С помощью бензойной кислоты учащимися может быть осуществлена имитация «зимнего пейзажа».
На картоне (с отверстиями) или на листе пористой (фильтровальной) бумаги монтируют небольшой макет сада. Под картоном (бумагой) нагревают в фарфоровой или железной чашке бензойную кислоту, накрыв всю установку стеклянным колпаком. Возгоняющаяся кислота, проходя через отверстия в картоне или поры в бумаге, осаждается на «деревьях» в виде красивого инея. Следует остерегаться попадания паров бензойной кислоты в атмосферу.
Получение бензойной кислоты окислением бензальдегида. Бензальдегид легко окисляется в бензойную кислоту кислородом воздуха.
На стеклянную пластинку, часовое стекло или в фарфоровую чашку помещают несколько капель бензальдегида. Через несколько минут начинают появляться кристаллы. К концу урока весь альдегид превращается в кислоту.
Взяв для опыта несколько миллилитров альдегида, таким способом можно получить бензойную кислоту в количестве, необходимом для демонстрации опытов с ней.
Получение бензола из бензойной кислоты (см. опыт на с. 63).
§
Свойства молочной кислоты. Молочная кислота является наиболее доступным и практически важным представителем оксикислот. Как известно, она образуется при скисании молока (в результате молочнокислого брожения лактозы), в процессе изготовления разнообразных молочнокислых продуктов, при силосовании кормов.
При нагревании с серной кислотой молочная кислота разлагается на уксусный ангидрид и муравьиную кислоту. Последняя в условиях опыта может разлагаться с образованием оксида углерода (II), который нетрудно обнаружить по характеру горения:
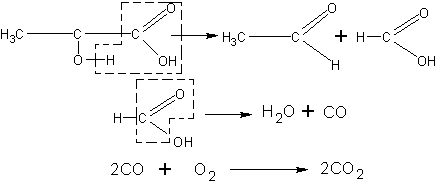
В пробирку наливают 1 мл молочной кислоты и двойное количество концентрированной серной кислоты. Пробирку закрывают пробкой с оттянутой на конце стеклянной трубкой. Нагревают смесь веществ (в вытяжном шкафу!) до кипения и поджигают выделяющийся газ. Он горит характерным для угарного газа синеватым пламенем.
При отсутствии нагревания выделение оксида углерода (II) прекращается.
Опыты с салициловой кислотой. Салициловая кислота представляет собой ортооксибензойную кислоту.
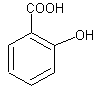
Знакомство с салициловой кислотой как веществом с двумя функциональными группами представляет значительный интерес.
а) Убеждаются в действии на лакмус водного раствора салициловой кислоты (получаемого при нагревании).
б) К раствору кислоты добавляют несколько капель раствора хлорида железа (III). Как и в случае фенола, образуется фиолетовая окраска.
в) При значительном нагревании кислота, подобно другим карбоновым кислотам, разлагается, выделяя углекислый газ:
Нагревают в пробирке с отводной трубкой кристаллы салициловой кислоты. Выделяющийся газ пропускают через известковую воду, образуется муть. Остаток в пробирке обладает резким запахом фенола.
г) Подобно бензойной кислоте, салициловая кислота возгоняется. В небольшую пробирку помещают 3—4 кристаллика салициловой кислоты, При нагревании на стенках пробирки быстро образуется белый налет кислоты, который при дальнейшем нагревании постепенно перемещается вверх, как при возгонке йода.
ГЛАВА VIII
§
Основное содержание эксперимента по данной теме составляют реакции этерификации и омыления. Сравнительная ограниченность опытов объясняется тем, что из способов получения сложных эфиров в школе рассматривается только реакция между кислотой и спиртом, а из химических свойств — только гидролиз.
СЛОЖНЫЕ ЭФИРЫ
При демонстрации способа получении сложных эфиров образующийся продукт обычно собирается в виде слоя над жидкостью, что трудно наблюдать учащимся на расстоянии. Наиболее убедительный признак образования эфира — появление характерного запаха. Поэтому первое ознакомление с получением эфиров лучше всего провести в виде лабораторного опыта.
Чтобы иметь возможность познакомить учащихся с получением различных эфиров и сделать заключение о том, что реакция этерификации является общей для образования веществ данного класса, можно одновременно дать учащимся синтез разных эфиров (например, метилового, этилового эфиров уксусной кислоты, этилового эфира муравьиной кислоты и др.).
Синтез этилового эфира уксусной кислоты (этилацетата). Опыт можно поставить так, чтобы одновременно показать влияние катализатора (серной кислоты) на скорость процесса.
1. В пробирку наливают 0.5 мл концентрированной уксусной кислоты (эссенции) и равный объем этанола. Запах эфира не обнаруживается даже при подогревании смеси.
После этого в пробирку добавляют 0,5—1 мл концентрированной серной кислоты. Появляется запах эфира, усиливающийся при дальнейшем нагревании.
2. В широкую пробирку (колбочку) наливают по 3 мл спирта, уксусной кислоты и концентрированной серной кислоты. Пробирку закрывают пробкой с обратным холодильником для конденсации паров спирта и эфира и нагревают в стакане с кипящей водой в течение 3—5 мин. Затем выливают жидкость в пробирку с насыщенным раствором поваренной соли. Серная кислота, а также не прореагировавшие спирт и уксусная кислота растворяются, эфир же высаливается и образует верхний слой, при этом ощущается сильный запах. Переливают смесь в делительную воронку и отделяют эфирный слой.
3. В небольшую колбу с отводной трубкой наливают по 10 мл этанола и уксусной кислоты и 5 мл концентрированной серной кислоты. Соединяют колбу с нисходящим холодильником (желательно водяным, так как точка кипения эфира 77°С) и отгоняют на кипящей водяной бане образующийся сложный эфир (до уменьшения объема в колбе наполовину).
Реакция этерификации обратима, но в этих условиях равновесие сдвигается вправо вследствие улетучивания эфира.
СН3СООН НОС2Н5 à СН3СООС2Н5 Н2O
Вместе с эфиром частично перегоняется спирт и в некоторой степени уксусная кислота (обычно спирт для реакции берут в избытке по сравнению с теоретически рассчитанным количеством).
Добавляют немного воды к дистилляту и смесь взбалтывают. Кислота и спирт растворяются в воде, эфир собирается слоем сверху. Демонстрируют продукт в таком виде или после отделения от водного слоя с помощью делительной воронки.
. Получающийся эфир содержит еще некоторое количество спирта, кислоты и воды. Однако дальнейшая очистка его в условиях демонстрационного опыта нецелесообразна.
4. Чтобы убедительно показать учащимся отщепление воды oт молекул спирта и кислоты при этерификации, можно провести реакцию, взяв в качестве катализатора безводный сульфат меди. Посинение последнего укажет на образование воды в ходе реак-
 ции. Некоторая сложность проведения опыта связана с тем, что для реакции требуются безводные реагенты (кислота и спирт).
ции. Некоторая сложность проведения опыта связана с тем, что для реакции требуются безводные реагенты (кислота и спирт).
В колбочку (рис. 34) наливают равные объемы спирта и кислоты и помещают 1—2 г безводного сульфата меди. К колбе присоединяют обратный холодильник с хлоркальциевой трубкой для защиты от атмосферной влаги. При встряхивании колбы уже без нагревания начинается постепенное посинение сульфата меди, что указывает на начало реакции. При нагревании смеси на водяной бане в течение 10 мин соль довольно быстро синеет и образуется значительное количество эфира. Запах становится очень сильным при выливании смеси в насыщенный раствор хлорида натрия.
5. С целью получения чистого образца этилацетата опыт ставят следующим образом (рис. 35).
В круглодонную колбу с капельной воронкой и отводной трубкой наливают 10 мл спирта и 10 мл концентрированной серной кислоты. Колбу нагревают через асбестированную сетку, чтобы термометр, опущенный в жидкость, показывал примерно 120°С, и добавляют из капельной воронки смесь 20 мл спирта и 20 мл уксусной кислоты с той же скоростью, с какой отгоняется эфир.

Чтобы освободить дистиллят от примеси уксусной кислоты, его нейтрализуют, добавляя постепенно насыщенный раствор карбоната натрия, контролируя с помощью лакмусовой бумажки рН среды. Смесь при этом пенится, особенно вначале, от выделяющегося углекислого газа. Отделяют эфир от водного слоя с помощью делительной воронки, взбалтывают с 10—15 мл 50-процентного водного раствора хлорида кальция. Хлорид кальция образует со спиртом молекулярное соединение и тем самым очищает эфир. Снова на делительной воронке отделяют эфир, добавляют к нему безводный сульфат натрия для связывания воды и перегоняют на водяной бане при температуре 77°С.
Получение этилового эфира бензойной кислоты (этилбензоата). В данном случае процесс этерификации протекает согласно следующему уравнению реакции:
С6Н5СООН НОС2Н5 Û С6Н5СООС2Н5 Н2О
В пробирке растворяют 1 г бензойной кислоты в 3-4 мл спирта и добавляют 7—8 капель концентрированной серной кислоты. При нагревании ощущается мятный запах эфира. При выливании в воду эфир всплывает наверх.
Синтез аспирина. Уксусный ангидрид является сильным ацилирующим средством и часто используется для получения сложных эфиров.
Ацилируя им салициловую кислоту (по фенольному гидроксилу), можно получить широко известное лекарственное средство — аспирин:
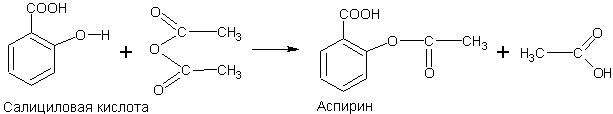
Как видно из формулы, аспирин одновременно является и ароматической кислотой, и сложным эфиром.
Для опыта взвешивают 1,3 г салициловой кислоты и 1,2 г уксусного ангидрида. Помещают вещества в небольшую колбочку, прибавляют к ним каплю концентрированной серной кислоты, за-
крывают колбу пробкой с обратным холодильником и нагревают смесь на водяной бане при 60°С в течение часа. Затем продолжают нагревание еще в течение часа при 90°С. После этого смесь в колбе охлаждают; отфильтровывают кристаллы аспирина (лучше это сделать на воронке Бюхнера) и промывают осадок ледяной водой. Для получения более чистого препарата необходимо дополнительно промыть его небольшим количеством холодного толуола. Сухой осадок взвешивают и определяют выход продукта по сравнению с теоретическим.
С аспирином можно проделать ряд опытов, но, разумеется, из-за недостаточной чистоты нельзя принимать его в качестве лекарства.
Гидролиз сложных эфиров. Опыты по гидролизу сложных эфиров расширяют общие представления учащихся о химической реакции, так как в весьма наглядной форме дополняют их сведениями об обратимости реакций и роли катализаторов.
Как известно, гидролиз катализируется минеральными кислотами, которые служат катализаторами и в реакции этерификации:
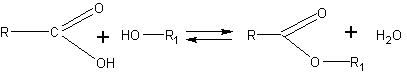
Не сдвигая равновесия в ту или другую сторону, минеральные кислоты значительно ускоряют наступление равновесия. Гидролиз эфиров в очень сильной степени ускоряется в присутствии щелочей, так как они связывают (превращают в соль, т.е. омыляют) образующуюся кислоту, вследствие чего реакция идет до конца в сторону разложения эфира.
а) В три пробирки помешают по нескольку капель этилацетата. В одну пробирку приливают воду в таком количестве, чтобы эфир только растворился в ней, в другую пробирку приливают раствор щелочи, в третью — раствор серной кислоты. Пробирки встряхивают и оставляют стоять или нагревают на водяной бане (в стакане с горячей водой). Через некоторое время убеждаются, что в пробирке со щелочью запах эфира быстро исчезает, в пробирке с кислотой он ослабевает, а в пробирке с водой остается без изменений.
В пробирке с кислотой удается обнаружить нары уксусной кислоты с помощью влажной лакмусовой бумажки (серная кислота нелетуча). В пробирке с водой не удается обнаружить кислоту даже в растворе (если препарат эфира был чистым).
б) В две пробирки помещают по 2 мл этилового эфира бензой-
ной кислоты. В одну пробирку приливают около 8 мл раствора серной кислоты (1 : 1), в другую — такой же объем воды. Нагревают пробирки в течение нескольких минут в пламени спиртовки. При охлаждении в первой пробирке выпадает осадок — кристаллы бензойной кислоты, во второй пробирке осадок не образуется, так как гидролиз не прошел.
в) В колбочку наливают 1 мл этилацетата и растворяют его в воде (около 20 мл). Убеждаются, что раствор (в отдельной пробе) не изменяет окраски лакмуса. Добавляют к раствору в колбе 2—3 капли фенолфталеина и очень немного раствора гидроксида натрия, чтобы только появилось не исчезающее при взбалтывании розовое окрашивание. Закрывают колбу пробкой с обратным холодильником и нагревают смесь в пламени горелки до кипения, после чего дают смеси остыть. Окраска фенолфталеина исчезает, так как щелочь нейтрализуется образующейся при гидролизе кислотой. Добавляют еще несколько капель щелочи и снова нагревают смесь до исчезновения окраски,
Прибавляя к образующемуся нейтральному раствору раствор хлорида железа (III), убеждаются в появлении уксусной кислоты при гидролизе эфира.
г) При длительном омылении эфира щелочью можно провести глубокий гидролиз и не только обнаружить, но и выделить образующиеся продукты.
Опыт можно дать для самостоятельной разработки учащимися.
Идея его такова. Если эфир кипятить со щелочью (25—30-процентной), взяв ее с некоторым избытком против количества, необходимого для нейтрализации образующейся кислоты, то через некоторое время эфирный слой вследствие гидролиза совершенно исчезнет. Чтобы эфир и спирт при этом не улетучивались (уксусная кислота будет в виде соли), кипячение следует вести в колбе с обратным холодильником. Пары спирта после окончания опыта .могут быть обнаружены по их горению. Кроме того, спирт можно отогнать и обнаружить иодоформной реакцией. Раствор ацетата натрия со щелочью можно обработать концентрированной серной кислотой, после чего отогнать уксусную кислоту.
Гидролиз аспирина. По месту сложноэфирной группировки аспирин может подвергаться гидролизу с образованием салициловой и уксусной кислот:
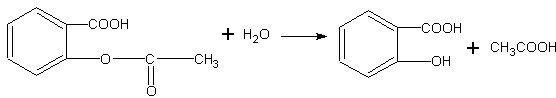
Салициловая кислота, содержащая в молекуле фенольный гидроксил, может быть обнаружена при помощи раствора хлорида железа (III).
Очень небольшое количество аспирина (примерно 0,1 г) растворяют в 4—5 мл воды. Часть раствора испытывают 1—2 каплями раствора хлорида железа. Характерной для фенолов окраски не наблюдается.
Другую часть раствора аспирина кипятят 2—3 мин и после охлаждения также испытывают хлоридом железа. В данном случае наблюдается характерное окрашивание, свидетельствующее о появлении в растворе салициловой кислоты (фенольного гидроксила).
Получение метилового эфира метакриловой кислоты (метилметакрилата) из органического стекла. Органическое стекло (плексиглас) широко применяется сейчас в технике и для изготовления многих предметов повседневного обихода (чернильницы, ручки, шкатулки, мундштуки, пуговицы и т.п.). Оно представляет собой полимер метилового эфира метилакриловой (метакриловой) кислоты
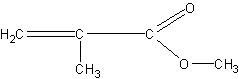
Строение полимера можно изобразить в виде следующей схемы:
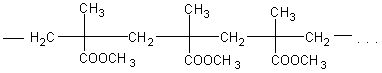
В предыдущих темах учащиеся познакомились со свойствами высокомолекулярных веществ и с их получением путем полимеризации и поликонденсации. На примере данного эфира можно их познакомить с разложением (деполимеризацией) высокомолекулярного продукта.
Опыт очень легко осуществляется и производит большое впечатление на учащихся.
В пробирку или колбочку с отводной трубкой и нисходящим воздушным холодильником насыпают стружки или мелкие кусочки органического стекла. Нагревая колбу через асбестовую сетку или на голом пламени, деполимеризуют органическое стекло и перегоняют образующийся при этом эфир. Перегонку можно производить без контроля термометром. До полного разложения всего органического стекла опыт лучше не доводить, так как продукт начинает обугливаться и колбу после опыта бывает трудно отмыть. В
приемнике собирается прозрачная, бесцветная жидкость с эфирным запахом — метилметакрилат.
Если эфир имеет слегка желтую окраску, его повторно перегоняют. отбирая фракцию в интервале 97-103°С (чистый эфир кипит при 100°С).
Получение полиметилметакрилата. Превращение метилового эфира метакриловой кислоты в высокомолекулярный продукт — полиметилметакрилат — наиболее доступный для школы пример реакции полимеризации.
Наливают в пробирку около 10 мл очищенного метилметакрилата. добавляют очень немного сухой перекиси бензоила (0,05 г) и встряхивают до растворения.
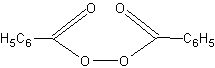
Затем реакционную смесь нагревают на водяной бане при 80— 85°С. Через 15-20 мин замечается увеличение вязкости жидкости. При дальнейшем нагревании жидкость постепенно превращается в твердую стекловидную массу. Пробирку разбивают и извлекают стерженек образовавшегося полимера. Можно получить окрашенный образец, если к исходному мономеру добавить небольшое количество органического красителя. Иногда полимеризацию метилового эфира метакриловой кислоты удастся наблюдать без применения катализатора при длительном стоянии жидкости.
Опыты с полиметилметакрилатом. Кроме деполимеризации с полиметилметакрилатом можно провести следующие опыты.
а) Нагревают над пламенем горелки или на асбестовой сетке пластинку из органического стекла, затем сгибают ее. Убеждаются, что полимер обладает свойством термопластичности.
б) Поджигают кусочек полиметилметакрилата. Он горит голубым некоптящим пламенем с характерным потрескиванием; ощущается эфирный запах.
ЖИРЫ
Школьные опыты по разделу жиров немногочисленны. Но так как жиры играют исключительную роль в жизни человека и учащиеся повседневно имеют с ними дело, следует стремиться к тому, чтобы на внеклассных занятиях и в виде домашних работ был поставлен с жирами целый комплекс работ практического значения.
Растворимость жиров. В 4 пробирки наливают по 1 мл воды, спирта, бензина и эфира и помещают примерно по одинаковому кусочку жира или по нескольку капель растительного масла. Про-
бирки встряхивают и наблюдают, что жир лучше всего растворяется в эфире. Пробирки, где растворение идет плохо, подогревают на горелке. Убеждаются, что в воде жир не растворяется даже при нагревании.
Несколько капель раствора жира в эфире и бензине наносят на фильтровальную бумагу. Наблюдают образование жирных пятен на бумаге после испарения растворителя.
Экстрагирование жиров и масел. Растворимостью жиров и масел в органических растворителях пользуются для извлечения их из отходов скотобоен, из семян, а также для удаления жировых пятен с одежды и т.д. В качестве растворителей используются, кроме указанных выше веществ, дихлорэтан С2Н4СI2, четыреххлористый углерод ССI4 и др.
а) В небольшую колбочку насыпают 5 г предварительно измельченных в ступке сухих семян льна или подсолнечника и наливают 10—15 мл бензина, дихлорэтана или четыреххлористого углерода.
Измельчение семян должно быть по возможности тщательным. С этой целью растирание можно производить, смешав семена с чистым речным песком. Колбу закрывают пробкой с обратным холодильником и кипятят на горелке через асбестовую сетку в течение 10—15 мин. Затем отфильтровывают раствор жира и наблюдают жировое пятно на бумаге после испарения растворителя. Образование небольшого количества масла можно наблюдать и при осторожном выпаривании из фарфоровой чашки.
б) Довольно простой способ извлечения масла состоит в следующем: 2-3 г тонко измельченных в ступке семян подсолнечника или льна помещают в колбу, приливают 25—30 мл диэтилового эфира и закрывают колбу корковой пробкой. Встряхивают колбу время от времени в течение часа. Полученный раствор масла в эфире фильтруют (при погашенных горелках). Остаток семян в колбе промывают дважды небольшими порциями эфира, которые затем также фильтруют и присоединяют к основному раствору. Эфир отгоняют из раствора на водяной бане.
Учащимся может быть предложено произвести необходимые взвешивания при проведении опыта и вычислить содержание масла в семенах.
в) В лаборатории для извлечения масел из семян часто пользуются специальными приборами — экстракторами. Экстрактор (рис. 36) состоит из трех частей: колбы, где нагревается растворитель, экстрактора, где происходит процесс растворения, и обратного холодильника, предназначенного для улавливания паров растворителя.
 Измельченные семена или другой материал, подвергающийся экстракции, помещают в гильзу из фильтровальной бумаги. Гильзу готовят, свертывая полоску фильтровальной бумаги в трубку и загибая края на одном конце внутрь трубки. В такую гильзу сначала помещают немного ваты, затем на нее насыпают материал, подвергаемый экстракции, снова кладут немного ваты и загибают внутрь верхние края гильзы. Гильзу помещают в экстрактор, а в колбу наливают растворитель (диэтиловый эфир) и герметично соединяют части аппарата. Нагревают колбу на водяной бане. Пары растворителя поднимаются в холодильник (обязательно заполненный водой) и конденсируются в нем. Жидкость из холодильника стекает в экстрактор, растворяет вещество и, заполнив экстрактор до определенного уровня, переливается по зигзагообразной сифонной трубке в колбу. За час кипячения растворителя может произойти 6—8 заполнений экстрактора и возвращений жидкости в колбу. Концентрация экстрагируемого вещества в колбе при этом все время возрастает. После отгонки растворителя получают нужный продукт.
Измельченные семена или другой материал, подвергающийся экстракции, помещают в гильзу из фильтровальной бумаги. Гильзу готовят, свертывая полоску фильтровальной бумаги в трубку и загибая края на одном конце внутрь трубки. В такую гильзу сначала помещают немного ваты, затем на нее насыпают материал, подвергаемый экстракции, снова кладут немного ваты и загибают внутрь верхние края гильзы. Гильзу помещают в экстрактор, а в колбу наливают растворитель (диэтиловый эфир) и герметично соединяют части аппарата. Нагревают колбу на водяной бане. Пары растворителя поднимаются в холодильник (обязательно заполненный водой) и конденсируются в нем. Жидкость из холодильника стекает в экстрактор, растворяет вещество и, заполнив экстрактор до определенного уровня, переливается по зигзагообразной сифонной трубке в колбу. За час кипячения растворителя может произойти 6—8 заполнений экстрактора и возвращений жидкости в колбу. Концентрация экстрагируемого вещества в колбе при этом все время возрастает. После отгонки растворителя получают нужный продукт.
Плавление и затвердевание жиров. Жиры представляют собой смесь различных веществ, преимущественно эфиров глицерина. Поэтому они, как и нефть, не имеют постоянной температуры кипения и плавления. Температурой плавления жира считается та конечная температура, при которой плавящийся жир превращается в прозрачную жидкость. Температурой застывания считается та максимальная температура, при которой происходит затвердевание жира.
Определение температуры плавления и затвердевания жира может быть выполнено в приборе, изображенном на рис. 37.
В пробирку помешают 2—5 г жира и закрывают пробкой с двумя отверстиями. Через одно отверстие проходит термометр, шарик которого находится в жире. Другое отверстие служит для выхода воздуха. Нагревают пробирку с жиром в стакане с водой. Наблюдают начало плавления жира, но в качестве температуры плавления отмечают ту температуру, при которой жир становится прозрачным.
Для определения температуры затвердевания в пробирку наливают растительное масло и охлаждают снегом или смесью снега с солью (в зависимости от сорта масла).
Чтобы охлаждение его было более равномерным, масло необходимо постоянно помешивать.
 Когда масло начнет затвердевать, понижение столбика ртути вначале прекращается, затем уровень ртути немного повышается, после чего снова начинает падать. Максимальную температуру отмечают как температуру застывания масла.
Когда масло начнет затвердевать, понижение столбика ртути вначале прекращается, затем уровень ртути немного повышается, после чего снова начинает падать. Максимальную температуру отмечают как температуру застывания масла.
Реакции непредельных жиров (масел). Растительные масла в своем составе содержат глицсриды непредельных кислот, таких как, олеиновая, линоленовая и др. Известно, что эти кислоты способны обесцвечивать бромную воду и раствор перманганата калия. Отметим, что аналогичным образом они ведут себя и в связанном состоянии, в виде жира.
а) В демонстрационную пробирку
наливают 5 мл жидкого масла (льняное, подсолнечное и др.) и концентрированную бромную воду (насыщенный раствор). При встряхивании пробирки бром обесцвечивается (белый фон!), образуется твердый продукт присоединения брома к глицериду. Таким же способом можно провести реакцию с раствором перманганата калия и испытать на непредельность другие масла (оливковое, кокосовое, рыбий жир).
Испытание различных масел с помощью перманганата калия может быть дано в качестве домашнего задания.
б) Кроме брома к непредельным жирам по месту двойной связи присоединяются хлор и иод. К 3—5 мл растительного масла добавляют 5 капель спиртового раствора йода. Встряхивают смесь и прибавляют к ней раствор крахмала. Синее окрашивание не образуется, так как иод вступил в реакцию с жиром. В контрольном опыте наблюдают синее окрашивание.
Реакцию с йодом используют на практике для характеристики жиров, так как по количеству расходуемого йода на насыщение двойных связей, по так называемому йодному числу, можно судить о степени непредельности жира.
Определение степени непредельности жиров. В непредельных углеводородных радикалах жиров может содержаться различное количество двойных связей. О степени непредельности жиров можно судить по количеству обесцвечиваемой ими бромной воды.
В пробирки помещают по 2-3 капли различных жиров (твердые жиры предварительно растапливают): свиного сала, коровьего масла, подсолнечного масла и др. Приливают к жирам по 1 мл четыреххлористого углерода или другого органического растворителя. Затем из бюретки добавляют в пробирки раствор брома до тех пор. пока окраска его не будет исчезать.
Сравнивают количество бромной воды, пошедшей на реакцию с разными жирами, и располагают жиры в ряд по возрастающей степени непредельности.
Определение содержания кислот в жирах. В жирах обычно всегда содержится некоторое количество свободных жирных кислот. Количество кислот возрастает при длительном хранении жиров вследствие частичного их разложения. С разложением жиров связано их прогоркание. О количестве кислот в жире можно сулить по количеству щелочи, идущей на нейтрализацию их в определенной навеске жира: по так называемому числу кислотности. Можно поставить два параллельных опыта: определение числа кислотности в свежем и прогорклом масле.
В конической колбочке растворяют 2 г животного масла в смеси 5 мл спирта и 5 мл эфира. К раствору добавляют 2 капли фенолфталеина и титруют 0,1 М раствором щелочи (добавляя его из бюретки по каплям) до появления розовой окраски, остающейся после встряхивания.
Вычисляют, сколько миллилитров раствора щелочи расходуется в том и другом случае на нейтрализацию 100 г жира. Убеждаются, что кислотное число прогорклого масла выше, чем свежего.
Омыление жиров. Получение мыла из жиров может быть выполнено в различных вариантах. Эти варианты отличаются друг от друга как по степени полноты гидролиза жира, так и по тому, в водной или спиртовой среде производится омыление. Производственный процесс осуществляется, как известно, в водной среде, но так как он идет довольно медленно, то на уроке омыление можно провести в спиртовом растворе. Спирт, обладающий свойством растворять не только щелочь, но и жир, создает гомогенную среду, что значительно ускоряет реакцию.
а) К 4—5 г топленого свиного сала или коровьего масла в колбочке приливают смесь 10 мл спирта, 5 мл воды и 2—3 г едкого кали. Колбочку закрывают пробкой с воздушным обратным холодильником (для конденсации паров спирта) и нагревают на горелке, время от времени перемешивая жидкость. Примерно через 10 мин кипячения омыление заканчивается, так как проба целиком растворяется в горячей дистиллированной воде (отсутствие жира). Часть полученного раствора выливают в стаканчик с 20—25 мл насыщен-
ного раствора поваренной соли. На поверхности выделяется (высаливается) мыло, которое собирают, отжимают в тряпке и используют для опытов (растворение в воде, образование пены, выделение жирных кислот при действии на него минеральных кислот). К другой части раствора прибавляют соляную кислоту. Выделяются нерастворимые в воде жирные кислоты.
б) В большой фарфоровой чашке расплавляют 10 г свиного или говяжьего жира и прибавляют 30 мл 30—35-процентного водного раствора щелочи. Смесь кипятят в пламени горелки, помешивая стеклянной палочкой, остерегаясь разбрызгивания. По мере выкипания в чашку добавляют воду. Минут через 10-15 пипеткой отбирают немного жидкости в пробирку с горячей водой. При взбалтывании получается непрозрачный раствор (эмульсия), так как часть жира к этому времени еще не разложится. Нагревание продолжают до тех пор, пока проба будет полностью растворяться в воде, образуя обильную пену. Это наступает примерно через час или более. После этого раствор упаривают минут 10 до образования густой массы (клеевого мыла) и охлаждают.
Часть полученного раствора выливают в стакан с насыщенным раствором хлорида натрия. Мыло всплывает в виде хлопьев (высаливается). Его собирают, отмывают, если нужно, водой от щелочи, а затем сплавляют в однородную массу. Пробуют мыть им руки и выполняют опыты, описанные ранее (с. 126).
Часть раствора обрабатывают соляной кислотой до выделения слоя жирных кислот. Если жидкость охладилась, кислоты выделяются в виде твердого слоя. Отфильтровывают осадок кислот, а фильтрат нейтрализуют осторожно содой и выпаривают медленно на водяной бане (не на голом огне, так как при сильном нагревании глицерин улетучивается с парами воды). Получают сиропообразный раствор глицерина. До полного удаления воды процесс можно не доводить.
Неудовлетворительные результаты опыта бывают иногда из-за того, что у учащихся не хватает терпения провести гидролиз до конца, и поэтому вместо мыла они иногда извлекают неразложившееся сало и, конечно, получают совершенно отрицательный результат при попытке вымыть этим продуктом руки.
В качестве классной демонстрации этот опыт может быть осуществлен в следующих вариантах:
1. Готовят смесь веществ для опыта, как сказано выше, и начинают ее нагревать. После этого сразу же переходят к опыту, начатому учащимися на внеклассных занятиях, в котором омыление жира доведено почти до конца. Заканчивают омыление, как указано выше, извлекают полученные продукты, устанавливают их природу.
2. Если урок химии двухчасовой, то начинают опыт, как указано выше, и поручают вести его подготовленному для этой цели ученику. Пока опыт идет, в классе разбираются следующие вопросы урока (например, получение стеарина, мыла и глицерина в промышленности, гидрогенизация жиров, проблема синтеза жиров и т.п.). Когда гидролиз в опыте закончится, внимание учащихся снова привлекается к нему, и последующие операции по выделению и исследованию продуктов служат одновременно средством повторения и закрепления материала.
ГЛАВА IX
УГЛЕВОДЫ
Тема «Углеводы» по насыщенности экспериментом занимает одно из первых мест в курсе органической химии. Здесь могут быть ярко показаны свойства веществ, играющих исключительную роль в жизни человека; здесь находят практическое использование ранее полученные учащимися знания (распознавание многоатомных спиртов, альдегидов и т.п.), дополняется экспериментальный материал предыдущих тем (получение спирта из полисахаридов), раскрываются важные процессы современной химической промышленности (гидролиз древесины, получение искусственного шелка и др.).
ГЛЮКОЗА
При изучении глюкозы преимущественное значение имеют опыты, иллюстрирующие ее химические свойства и обосновывающие ее строение. На примере глюкозы впервые дается вывод структурной формулы полифункционального соединения.
Если при изучении спиртов не демонстрировалось брожение глюкозы или сахарозы, этот опыт ставится в данном разделе.
Получение глюкозы гидролизом полисахаридов целесообразно рассмотреть позднее в связи с изучением химических свойств крахмала и клетчатки, когда процесс может быть лучше понят учащимися.
Физические свойства глюкозы. Учащиеся предварительно знакомятся с внешним видом и вкусом глюкозы.
а) Растворяют глюкозу в пробирке с водой до получения насыщенного раствора. Затем растворяют дополнительное количество ее при нагревании. Отмечают, сильно ли влияет нагревание на изменение растворимости глюкозы.
б) В двух пробирках растворяют в равных объемах воды по 5 г глюкозы и обыкновенного сахара (сахарозы). Убеждаются, что раствор сахарозы имеет более сладкий вкус.
Реакции спиртовых групп глюкозы. При ознакомлении с молекулярной формулой глюкозы С6Н12О6 внимание учащихся может быть обращено на значительное содержание кислорода в этом веществе. Учащимся уже известен ряд кислородсодержащих органических веществ (спирты, альдегиды, кислоты и др.). Поэтому может быть поставлен вопрос: не содержит ли глюкоза ранее изученные функциональные группировки атомов? Испытывая раствор глюкозы лакмусом, убеждаются, что глюкоза не является кислотой.
Исходя из наличия большого количества атомов кислорода в молекуле, можно высказать предположение о принадлежности глюкозы к многоатомным спиртам и проверить это предположение реакцией с гидроксидом меди (II), известной учащимся по многоатомному спирту — глицерину.
Так как гидроксид натрия в этой реакции должен быть в избытке (с. 90), то опыт лучше продемонстрировать в следующем порядке.
В демонстрационной пробирке или в стакане к 50 мл 10-процентного раствора глюкозы приливают около 10 мл раствора едкого натра и по каплям добавляют раствор сульфата меди. При взбалтывании смеси наблюдают образование темно-синей окраски — результат взаимодействия гидроксида меди (II) с глюкозой. Чтобы учащиеся сознательно отнеслись к этому опыту, следует с ними вспомнить предварительно образование глицерата меди.
Дальнейшим шагом в изучении строения глюкозы было бы определение числа гидроксильных групп в молекуле. Однако экспериментальное решение этого вопроса в школьных условиях затруднительно. Поэтому учитель лишь сообщает, что число гидроксильных групп в молекуле глюкозы может быть выяснено на основе изучения реакции этерификации. Известен, например, пентауксусный эфир глюкозы*. Поскольку уксусная кислота одноосновна, то очевидно, что в молекуле глюкозы содержится пять гидроксильных групп. Так как гидроксильные группы (что известно учащимся) могут находиться лишь у разных атомов углерода, то делается заключение о том, что с ними связано 5 углеродных атомов в молекуле глюкозы.
Дальнейшая задача будет заключаться в том, чтобы установить, какая еще функциональная группа находится у шестого атома углерода. Поскольку это не гидроксильная (спиртовая) и не карбоксильная (кислотная) группа, то можно предположить: не является ли она альдегидной?
Реакция альдегидной группы. Учащимся известна характерная реакция альдегидов — реакция серебряного зеркала. Этой реакцией и пользуются, чтобы проверить, не является ли глюкоза альдегидом, одновременно с принадлежностью ее к многоатомным спиртам.
а) В хорошо вымытую (щелочью, а затем хромовой смесью и водой) колбочку наливают 20 мл аммиачного раствора гидроксида серебра и прибавляют 10 мл 10-процентного раствора глюкозы. Смесь слегка нагревают, держа колбочку над пламенем горелки или поместив ее в стакан с горячей водой. Быстро образуется ровный блестящий слой серебра. При добавлении раствора глюкозы к аммиачному раствору гилроксида серебра вначале образуется темный осадок. Он не помешает появлению зеркала при нагревании смеси.
Опыт с несомненностью устанавливает наличие альдегидной группы атомов в молекуле глюкозы.
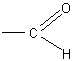
Если принять теперь во внимание, что атомы углерода составляют прямую цепь, то расшифрованную часть молекулы можно будет изобразить в таком виде:
Оставшиеся 6 атомов водорода присоединены, очевидно, за счет неиспользованных валентностей атомов углерода.
б). Реакция с гидроксидом меди (II). Если со второй характерной реакцией альдегидов — восстановлением гидроксида меди — учащиеся ранее не были ознакомлены (с. 101), то ее необходимо рассмотреть здесь. Она потребуется в дальнейшем при определении продуктов гидролиза полисахаридов. При нагревании альдегидов с гидроксидом меди (11) прежде всего образуется желтый осадок гидроксида меди (I), который при дальнейшем нагревании превращается в красный оксид меди (I).
В демонстрационной пробирке к 10—15 мл 10-процентного раствора глюкозы приливают равный объем разбавленной щелочи и 1 мл разбавленного раствора сульфата меди (II). Нагревают в пламени спиртовки образовавшийся темно-синий раствор. Наблюдают образование желтого, а затем красного осадка.
Чтобы образование красного осадка было отчетливым, не следует пользоваться концентрированными растворами щелочи и медного купороса и нельзя приливать сульфата меди слишком много (должен быть избыток щелочи). При несоблюдении этих условий вместо красного осадка иногда образуется желтый или бурый осадок (вследствие частичного образования черного оксида меди (II)).
Доказательство принадлежности глюкозы одновременно к многоатомным спиртам и альдегидам может быть проведено в одном опыте. Такой опыт требует очень немного времени и уместен при обобщающих ответах учащихся и на практических занятиях.
К раствору глюкозы приливают раствор щелочи и, как было описано выше, очень немного раствора сульфата меди. Появляется темно-синее окрашивание, что свидетельствует о принадлежности глюкозы к многоатомным спиртам. При нагревании раствора сначала образуется желтый, а затем красным осадок оксида меди (I), что указывает на наличие в глюкозе и альдегидной группы.
Обнаружение глюкозы во фруктах и ягодах. С помощью реакции восстановления гидроксидом меди Сu(ОН)2 проверяют наличие глюкозы в виноградном соке, цветах клевера, меде, соке малины, в спелых яблоках и т.п.
Брожение глюкозы. Демонстрация брожения глюкозы необходима в том случае, если соответствующий опыт не ставился при изучении спиртов. Опыт выполняют, пользуясь приведенным ранее описанием (с. 82).
САХАРОЗА
Изменение сахара при нагревании. Опыты нагревания сахара могут быть поставлены как в виде демонстрации, так и в виде домашней работы учащихся.
Нагревают в пробирке или фарфоровой чашке толченый сахар до плавления (160°С). Выливают часть расплава на бумагу. По мере остывании образуется леденец.
При более сильном нагревании (до 200°С и выше) сахар окрашивается в желтый цвет, при этом образуется карамель. Если са
хар нагревают далее, он еще более темнеет и наконец обугливается, выделяя белый дым: сухая перегонка. Выделяющиеся газообразные продукты можно поджечь. Легко можно составить коллекцию разнообразных продуктов, образующихся при нагревании сахара. При всяком изменении окраски часть расплава отливают в маленькую пробирочку, где он и застывает. Пробирочки монтируют на щитке в порядке углубления окраски сахара.
Обугливание сахара концентрированной серной кислотой. Серная кислота, отнимая у сахара молекулы воды и частично окисляя его, освобождает чистый углерод.
В небольшом химическом стакане смачивают водой 10 г толченого сахара и прибавляют равный объем конце<<script async src=»https://pagead2.googlesyndication.com/pagead/js/adsbygoogle.js?client=ca-pub-2375543522969552″
crossorigin=»anonymous»>
§
Основная идея опытов по данному разделу — показать разнообразные применения клетчатки, основанные на использовании ее химических свойств. В связи с этим опыты подразделяются па три основные группы: а) гидролиз клетчатки, вскрывающий принадлежность ее к классу углеводов и иллюстрирующий основной процесс современной гидролизной промышленности; б) нитрование клетчатки, показывающее использование ее для производства взрывчатых веществ.
Из сказанного видно, что материал данного раздела имеет существенное значение для практического ознакомления учащихся с научными основами современных химических производств.
Гидролиз клетчатки до глюкозы. Одним из характерных и практически важных свойств клетчатки как представителя полисахаридов является ее гидролиз до более простых продуктов.
Конечным продуктом гидролиза клетчатки, как и крахмала, является глюкоза:
(С6Н12O6)n nH2O à С6Н12O6
Гидролиз клетчатки также катализируется минеральными кислотами.
В настоящее время в промышленности гидролизом клетчатки (преимущественно древесины) и последующим сбраживанием образующейся глюкозы получают этиловый спирт и другие продукты.
Опыты по гидролизу могут ставиться с различной полнотой: от констатации наличия глюкозы в гидролизате до извлечения глюкозы в твердом виде.
а) В ступке смачивают кусочек чистой фильтровальной бумаги или ваты концентрированной серной кислотой и растирают пестиком. Полученный раствор переносят в демонстрационную пробирку или химический стакан с небольшим количеством воды (2-3 мл) и кипятят в течение 5 мин. После этого часть раствора нейтрализуют щелочью и нагревают с фелинговой жидкостью. Образование красного оксида меди (I) указывает на появление глюкозы.
Параллельно можно провести контрольный опыт: кипятить столько же времени фильтровальную бумагу или вату с водой без кислоты. При последующем нагревании этой жидкости с фелинговым раствором осадок оксида меди (I) не образуется.
б) Для демонстрации процесса гидролиза клетчатки можно воспользоваться древесными опилками. Этот опыт будет поучительнее предыдущего, так как с большим приближением иллюстрирует современное гидролизное производство.
1—2 г древесных опилок смачивают в химическом стакане водой, прибавляют 2—3 мл концентрированной серной кислоты, затем 30—50 мл воды и кипятят 8—10 мин. После этого часть еще горячего раствора фильтруют через складчатый фильтр, фильтрат нейтрализуют щелочью и нагревают с фелинговым раствором. Образуется красный оксид меди (1).
Параллельно может быть поставлен опыт нагревания опилок с водой без кислоты. Получающийся в этом случае раствор не дает с фелинговой жидкостью оксид меди (I).
Время, в течение которого идет кипячение смеси опилок с кислотой, может быть использовано для разъяснения учащимся сущности гидролизного производства и его значения.
в) Предыдущие опыты дают представление о реакции гидролиза клетчатки и о получаемом продукте, но еще не знакомят с основными стадиями гидролизного производства. Как показала практика, в школьных условиях можно поставить опыт, более полно отражающий промышленный способ осахаривания древесины.
Ввиду длительного времени некоторые операции опыта (нейтрализация кислоты и фильтрование) можно выполнить вне урока.
Наиболее распространенный в промышленности способ осахаривания древесины состоит в нагревании ее с очень разбавленной серной кислотой (0,5%) под давлением до 10 ат. В школе, естественно, нельзя поставить опыт с применением автоклава, да в этом и нет особой необходимости, так как не ставится задача получения максимально возможного выхода глюкозы. Зато можно несколько повысить концентрацию серной кислоты, чтобы ускорить процесс.
К 1-1,5 г древесных опилок в колбе прибавляют 14 мл серной кислоты (6 мл воды и 8 мл концентрированной серной кислоты). Смесь встряхивают примерно в течение 10 мин, одновременно прогревая ее немного в пламени спиртовки. Затем к ней приливают 150—200 мл воды, закрывают колбу пробкой с обратным холодильником и кипятят в течение 10-15 мин.
Раствор фильтруют в большую коническую колбу. К фильтрату добавляют небольшими порциями тонко измельченный мел, все время энергично круговыми движениями перемешивая смесь до тех пор, пока не прекратится выделение углекислого газа (шипение) и лакмусовая бумажка не покажет, что серная кислота нейтрализована:
СаСО3 H2SO4 à- CaSO4 Н2О СО2
Вместо мела можно применить известковое молоко или баритовую воду.
Раствор фильтруют от обильного осадка сульфата кальция. С помощью фелингова раствора убеждаются в наличии глюкозы в фильтрате. Упаривают раствор в фарфоровой чашке на водяной бане до небольшого объема жидкости.
Так как фильтрование и упаривание занимают много времени, то эта часть опыта может быть поставлена в двух вариантах: учащимся сообщается, что эти операции будут выполнены после урока и на следующем занятии из концентрированного раствора будет извлечен сахар, или же берется упаренный раствор, заранее подготовленный учащимися в кружке. Второй путь более удобный, так как не нарушает логической стройности урока и ведет к лучшим результатам. Последнее объясняется тем, что в процессе внеклассной работы учащиеся могут продолжительное время обрабатывать опилки слабо разбавленной кислотой (от 2 ч до суток) и длительное время кипятить их с сильно разбавленной кислотой (до 2 ч), что приведет к большему выходу продукта.
Сконцентрированный раствор глюкозы выпаривают в фарфоровой чашке на водяной бане. При этом следят за тем, чтобы выделяющийся сахар по возможности не подгорал. На дне чашки по мере испарения воды образуется немного глюкозы, обычно светло-желтого цвета (вследствие частичного разложения органических примесей). Полученный продукт можно испробовать на вкус — учащиеся убеждаются в его сладком вкусе. В классе ощущается запах горелого сахара, так как при выпаривании без вакуума не удается избежать частичного разложения. Растворив полученный продукт в воде и добавив дрожжей, в соответствующих условиях можно наблюдать реакцию брожения (по выделению углекислого газа), что подтверждает образование сахара из древесины.
Нитрование клетчатки и опыты с нитроклетчаткой. В зависимости от концентрации применяемой азотной кислоты и времени нитрования получаются, как известно, различные продукты. Для полного нитрования клетчатки в соответствии с уравнением нужна дымящая азотная кислота (уд. вес 1,52), которой обычно школы не располагают.
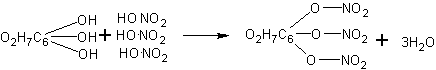
В случае применения азотной кислоты удельного веса 1,4 нитрование обычно не доходит до образования тринитроклетчатки. Однако с полученным продуктом удается проделать все необходимые опыты, иллюстрирующие свойства нитроклетчатки.
В химическом стаканчике готовят смесь 5 мл концентрирован-
ной азотной кислоты (уд. вес 1,4) и 10 мл концентрированной серной кислоты (уд. вес 1,84). После того как смесь примет комнатную температуру, в нее погружают минут на 10—15 комочек ваты, перемешивая его с кислотами стеклянной палочкой. Затем вату промывают водой в большом химическом стакане и в струе под водопроводным краном. После этого нитроклетчатку отжимают и высушивают в листах фильтровальной бумаги, неоднократно меняя их и разрыхляя каждый раз вату.
Полного высушивания нитрованной ваты на уроке не достичь; однако дальнейшие опыты удаются и со слегка влажной нитроклетчаткой. Можно также взять для опытов ранее заготовленную нитроклетчатку.
а) Горение нитроклетчатки. В пламени спиртовки поджигают одновременно небольшой комочек ваты и полученного нитропродукта (взятые в щипцы). Первый образец горит медленно, второй сгорает моментально.
Головку одной спички обертывают ватой, другой спички — нитроклетчаткой. Вату и нитроклетчатку поджигают. Вслед за горением ваты спичка вспыхивает, нитроклетчатка же сгорает настолько быстро, что спичка не успевает воспламениться.
б) Получение бездымного пороха. Немного нитроклетчатки в стакане смачивают этилацетатом или ацетоном до образования студенистой массы. После высыхания образуется бездымный порох. Извлекают часть его и поджигают. Горение происходит менее интенсивно, чем у нитроклетчатки.
АМИНЫ. КРАСИТЕЛИ
Изучению аминов в курсе органической химии предшествует изучение нитросоединений. Оно проводится преимущественно в плане обобщения, так как с отдельными представителями этого класса веществ учащиеся знакомились ранее. В связи с этим опыты для изучения нитробензола, пикриновой кислоты и других нитросоединений описаны в предыдущих главах и здесь не приводятся.
Опыты с алифатическими (жирными) аминами ниже даны сравнительно в небольшом количестве, так как эти соединения совершенно не рассматриваются в курсе химии или о них сообщаются лишь краткие сведения.
Красители, даже самые простейшие, обычно не изучаются в школе. Однако, учитывая большое образовательное значение знакомства с ними и высокий интерес учащихся к синтетической химии, ниже мы даем серию опытов для внеклассной работы
ЖИРНЫЕ АМИНЫ
Основная идея опытов с жирными аминами заключается в том чтобы представить амины как органические основания
В соответствии с этим наиболее важными Являются опыты окрашивания лакмуса водным раствором аминов; реакция солеобразования с кислотами; реакция выделения аминов из солей действием щелочей. Уяснение этих явлений позволит учащимся понять в дальнейшем свойства важнейшего представителя ароматических аминов — анилина.
Методическая сложность проведения опытов заключается в том, что начинать их приходится с получения аминов, чтобы иметь возможность далее демонстрировать их свойства Реакция же получения аминов действием щелочей на их соли не может быть понята учащимися до тех пор, пока они не познакомятся со свойствами этих соединений.
Поэтому при получении аминов на уроке тем или иным способом химизм образования их не рассматривают, а сразу переходят к изучению свойств. Возвращаются же к химизму лишь тогда, когда будет показано, что амины образуют с кислотами coли (например CH3NH2 HCI à CH3NH3CI) и могут быть выделены из последних щелочами (CН3NH2•HCI NaOH à СН3 NH NaCI Н2О)
Получение аминов из селедочного рассола. В селедочном рассоле содержится значительное количество аминов, преимущественно диметиламина (CH3)2NH и триметиламина (СН3)3N. Их присутствием и обусловливается характерный запах рассола
При отсутствии хлористоводородной соли метиламина вполне можно воспользоваться селедочным рассолом для практического ознакомления со свойствами жирных аминов
а) В большую круглодонную колбу с отводной трубкой наливают 15-20 мл селедочного рассола, добавляют 6-7 мл концентрированного раствора гидроксида натрия (раствор должен быть сильно щелочным), нагревают и отгоняют амины в пробирку с водой, охлаждаемую снегом или льдом. Через несколько минут образуется раствор, с которым можно проделать следующие опыты иллюстрирующие основные свойства аминов.
К части раствора приливают нейтральный раствор лакмуса или раствор фенолфталеина. Лакмус синеет, фенол-фталеин становится
малиновым. Делается вывод о том, что продукт соединения аминов с водой (рассматривать можно на примере метиламина) обладает щелочными свойствами, подобно раствору аммиака:
NН3 НОН à NH4OH
CH3NH2 НОН à CH3NH3OH
К части раствора приливают раствор хлорида железа (III). Параллельно ставят опыт взаимодействия хлорида железа (III) с раствором аммиака. Наблюдают образование одинаковых осадков, что подтверждает основные свойства водного раствора амина:
FeCI3 3NH4OH à Fe(OH)3 3NH4CI
FeCl3 3CH3NH3OH à Fe(OH)3 3CH3NH3CI
Кипятят в пробирке немного полученного раствора с добавкой щелочи и к выделяющимся парам подносят лучинку, смоченную концентрированной соляной кислотой. Образуется белый дым хлорида метиламмония (черный фон!).
Предыдущие опыты показывают большое сходство аминов с аммиаком. Для установления отличия их от аммиака и принадлежности к органическим соединениям демонстрируют горючесть аминов. Кипятят раствор амина и поджигают пары его у отверстия пробирки.
Последние два опыта получаются лишь в том случае, если, пользуясь селедочным рассолом, удается получить не слишком слабый раствор аминов.
б) Нагревают в колбе смесь селедочного рассола со щелочью, как было указано выше. К отводной трубке колбы подносят влажную лакмусовую бумажку, бумажка синеет. Подносят стеклянную палочку, смоченную концентрированной НСI, образуется белое облачко хлоридов аминов. Поджигают пары аминов у отводной трубки, они горят бледным пламенем. Пропускают пары в раствор хлорида железа (III), образуется осадок гидроксида железа (III).
Получение метиламина из хлористоводородной соли и опыты с ним. Предварительно учащиеся знакомятся с запахом метиламина. Для этого в пробирке или на стекле к небольшой щепотке хлорида метиламина они прибавляют 5—7 капель концентрированного раствора щелочи. Отмечают сходство запаха выделяющегося метиламина с запахом аммиака.
Для демонстрации опытов с метиламином в небольшую колбочку с отводной трубкой и капельной воронкой помещают 2—3 г соли, в капельную воронку наливают концентрированный раствор щелочи. Метиламин получают, прибавляя понемногу из воронки щелочь. С метиламином проделывают следующие опыты:
а) Пропускают выделяющийся газ в воду, подкрашенную фиолетовым или красным лакмусом. Лакмус синеет. Делают заключение о том, что не только по запаху метиламин имеет сходство с аммиаком, но и по химическим свойствам (щелочные свойства водного раствора). Составляется уравнение.
б) Получают снова раствор метиламина, пропуская газ в воду (без лакмуса), и проводят реакцию с раствором хлорного железа, как было указано выше. Подтверждаются щелочные свойства гидроксида метиламмония.
в) К отводной трубке колбы подносят открытую склянку с концентрированной соляной кислотой или палочку, смоченную кислотой. Образуется густое облако дыма хлористоводородной соли (черный фон!). Параллельно ставят опыт с аммиаком. Составляют уравнение реакции солеобразования и, проводя далее аналогию с аммиаком и солями аммония, объясняют реакцию получения амина из соли (см. выше).
г) Чтобы показать отличие метиламина от аммиака, демонстрируют горючесть метиламина, поджигая газ у отводной трубки колбы. Учащиеся знают, что аммиак на воздухе не горит. Для большей убедительности над пламенем горящего метиламина помещают стакан, сполоснутый известковой или баритовой водой. Появление мути на стенках свидетельствует о наличии углерода в горящем веществе (чего нет в аммиаке) и о принадлежности его к органическим соединениям.
§
Опыты с анилином должны показать, что это вещество принадлежит к классу аминов, имеет свойства основания и в то же время отличается от алифатических (жирных) аминов по степени проявления основных свойств. Большое внимание также должно быть уделено получению анилина из нитробензола ввиду особого исторического значения этой реакции и ее роли в современной промышленности.
Постановка опыта получения анилина из нитробензола с выделением продукта требует длительного времени. Поэтому он может быть поставлен или с помощью приемов, рекомендуемых для демонстрации длительных опытов, или без выделения продукта. При этом ограничиваются констатацией появления нового вещества с помощью качественной реакции. Более полный опыт с извлечением анилина переносится на практические занятия.
Ввиду ядовитости паров анилина при работе с ним должны соблюдаться меры предосторожности. Опыты по возможности следует проводить в вытяжном шкафу.
Отношение анилина к индикаторам. После ознакомления с физическими свойствами, составом и строением анилина, естественно, встает вопрос о том, что это вещество должно иметь химические свойства, сходные со свойствами рассмотренных ранее жирных аминов. С целью проверки этого предположения исследуют отношение анилина к индикаторам.
К 100 мл воды прибавляют 1 мл анилина и взбалтывают смесь стеклянной палочкой. Половину раствора отливают в другой стакан и прибавляют в один из них фенолфталеин, а в другой — лакмус. Окраска индикаторов не изменяется.
Взаимодействие анилина с кислотами. После того как показано, что анилин не действует на индикаторы, т.е. не обладает основными свойствами, возникает вопрос, способен ли он к солеобразованию.
а) В пробирку с 5 мл воды учащиеся наливают около 0,3 мл анилина. Смесь взбалтывают. Убеждаются, что анилин почти не растворился в воде. Добавляют по каплям концентрированную соляную кислоту. Анилин растворяется. Составляют уравнение реакции образования соли.
К. раствору соли анилина добавляют по каплям концентрированный раствор щелочи. Снова выделяется анилин в виде маслянистой жидкости. Составляют уравнение реакции.
При проведении опыта следует пользоваться концентрированными растворами кислоты и щелочи с той целью, чтобы не вводить большого количества воды, иначе из-за частичной растворимости анилина (1 г анилина растворяется в 30 мл воды) выделение его будет малозаметно.
б) В демонстрационную пробирку наливают 5 мл анилина и затем примерно такой же объем концентрированной соляной кислоты или 50-процентной серной кислоты. Дают некоторым учащимся убедиться, что проходит экзотермическая реакция. Охлаждают пробирку в струе холодной воды. Выделяются кристаллы соли. Растворяют соль по возможности в небольшом количестве воды (воду добавляют небольшими порциями и пробирку встряхивают). Если остался непрореагировавший анилин, отфильтровывают от него раствор, а к раствору соли добавляют концентрированный раствор щелочи. Выделяется свободный анилин.
Тем или другим из этих опытов устанавливается, что анилин является основанием; отсутствие же действия его на индикаторы говорит о том, что он основание более слабое, чем жирные амины.
Так как по составу молекул жирные амины и анилин отличаются тем, что вместо предельного радикала здесь имеется бен-
зольное ядро, то очевидно, что ослабление основных свойств аминогруппы обусловлено влиянием ароматического кольца.
Взаимодействие анилина с бромной водой. Как известно, влияние атомов друг на друга в молекуле взаимно. Если бензольное кольцо влияет на аминогруппу в анилине, ослабляя проявление се свойств основания, то можно ожидать, что и бензольное ядро под влиянием этой группы будет обладать свойствами, отличными от свойств бензола. С подобным изменением свойств ядра учащиеся уже встречались на примере фенола.
Встает вопрос, не ослабляет ли аминогруппа связь атомов водорода с ядром, подобно тому как эту связь ослабляет гидроксильная группа в феноле, и не будет ли анилин реагировать с бромной водой, как реагирует с ней фенол.
В колбе или демонстрационной пробирке растворяют при встряхивании и 10 мл воды I мл анилина. К полученному раствору (эмульсии) добавляют бромную воду до образования белого осадка.
Осадок представляет собой триброманилин:
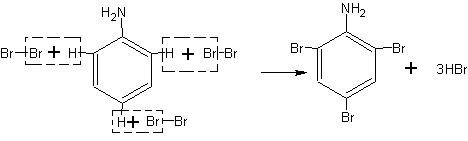
Для успеха опыта необходимо, чтобы растворы анилина и брома были достаточно концентрированными. Вместо раствора анилина можно воспользоваться раствором его соли.
Окисление анилина. Анилин сравнительно легко окисляется в разнообразные продукты. Окислением анилина, в частности, объясняется потемнение его при хранении в плохо закрываемых склянках. При окислении анилина хромовой смесью и некоторыми другими сильными окислителями образуется так называемый черный анилин, применяемый для окраски в черный цвет тканей, дерева, кожи.
а) В демонстрационной пробирке к 3-4 мл водной эмульсии анилина приливают 10—12 мл хромовой смеси. Жидкость взбалтывают и подогревают. Вначале она приобретает зеленую окраску. затем синеет, при дальнейшем нагревании образуется осадок черного анилина. Характер осадка выявляется при сильном взбалтывании смеси в пробирке.
б) В 100 мл воды растворяют 2 г двухромовокислого калия К2Сг2О7 и 1 мл концентрированной серной кислоты. К полученному раствору добавляют 2 г хлорида анилина или по 1 мл анили-
на и концентрированной соляной кислоты. Смесь перемешивают стеклянной палочкой. В образующийся раствор быстро вносят кусок хлопчатобумажной ткани. Краситель с тканью нагревают, затем ткань достают, отжимают валиком на гладкой поверхности и развешивают на стеклянных палочках.
в) Готовят раствор хлорида анилина, приливая к 50 мл воды 2 мл анилина и 3 мл концентрированной соляной кислоты. Затем к реакционной смеси добавляют еще 1 г хлорида аммония. Полученный раствор наносят кисточкой на гладкую поверхность доски.
После того как раствор впитается, таким же способом покрывают дерево вторым раствором, приготовляемым растворением 4 г бертолетовой соли и 6 г медного купороса в 50 мл воды. Бертолетова соль окисляет анилин, и поверхность доски постепенно приобретает черную окраску. Хлорид аммония и сульфат меди способствуют ускорению процесса окисления и получению плотной и прочной окраски. После высыхания поверхности на нее еще несколько раз наносят попеременно первый и второй растворы.
Получение анилина. Производство анилина долгое время было основано только на восстановлении нитробензола. Эта реакция была открыта знаменитым русским ученым Н.И.Зиминым.
В промышленном способе производства для восстановления нитробензола используются соляная кислота и железные или чугунные опилки. Все эти вещества загружаются в реактор, образующийся водород в момент выделения энергично восстанавливает нитробензол в анилин.
В школьных опытах получения анилина для ускорения реакции вместо железа используют обычно олово или цинк. Соображения экономического характера при таких незначительных масштабах здесь не играют роли. Опыт с железом может быть перенесен на практические занятия или в химический кружок и поставлен там не только для иллюстрации основной химической реакции, но и с целью отражения важнейших производственных операций.
а) В колбочку емкостью 100 мл вносят 2-3 капли нитробензола, приливают 1—2 мл концентрированной соляной кислоты и вносят 2-3 маленьких кусочка цинка (или олова). Если реакция пойдет слишком бурно, колбочку охлаждают в струе холодной воды или в стакане с водой. Когда запах нитробензола исчезнет или почти исчезнет, в колбу приливают концентрированный раствор щелочи (30-40-процентной) до тех пор, пока образующийся вначале осадок гидроксида цинка Zn(OH)2 ввиду своей амфотерности не растворится при дальнейшем прибавлении щелочи:
Zn(OH)2 2NaOH à Na2ZnO2 2H2O
Это будет гарантией того, что щелочь выделит анилин из соли.
Чтобы констатировать образование анилина, присоединяют к колбе холодильник и отгоняют около 0,5-1 мл жидкости. Анилин гонится вместе с парами воды в виде белой эмульсии. Затем дистиллят разбавляют водой, чтобы анилин растворился, и открывают его с помощью бромной воды. Одновременно убеждаются, что нитробензол не взаимодействует с бромной водой.
б) Препарат анилина для классных занятий может быть получен в опыте, более точно воспроизводящем промышленный способ, чем предыдущие.
В заводской практике для восстановления нитробензола берут значительно меньше соляной кислоты, чем соответствует уравнению
С6Н5NО2 6Н à C6H5NH2 2Н2О,
так как образующийся хлорид железа катализирует реакцию:
С6Н5NO2 2Fe 4Н2O à 2Fe(OH)3 C6H5NH2
В условиях школьного опыта, при работе с меньшими массами веществ, лучше брать количества, близкие к теоретическим.
В большую круглодонную колбу помещают 15—20 г нитробензола, 50 г железных опилок или мелких стружек и приливают небольшими порциями (сначала по 3-5 мл) 80—100 мл концентрированной соляной кислоты. После каждой добавки кислоты колбу закрывают пробкой с обратным воздушным холодильником для конденсации паров нитробензола и встряхивают. В случае слишком бурного течения реакции колбу непродолжительное время охлаждают в холодной воде. После того как будет введена вся кислота и реакция через некоторое время начнет затихать, колбу в течение 20—30 мин подогревают на асбестовой сетке. По исчезновению в колбе запаха нитробензола судят о полном восстановлении. Этот процесс требует обычно не менее часа.
Для выделения анилина из соли жидкость переливают в колбу для перегонки, добавляют к раствору понемногу концентрированной щелочи. При этом образуется осадок гидроксида железа (III). Прибавлять щелочь следует до сильнощелочной реакции (проба лакмусовой бумажкой), тогда избыток щелочи выделит анилин из соли.
Собирают прибор для перегонки с водяным паром (рис. 38).
Чтобы вода в парообразователе кипела без толчков, его снабжают предохранительной трубкой, доходящей до дна. Одновременно с парообразователем нагревают и колбу с анилином, чтобы поступающие пары воды в ней не конденсировались. Отгоняющийся анилин образует в приемнике водную эмульсию и частично соби-
рается в виде маслянистого слоя. Перегонку ведут до тех пор, пока в приемник не начнут падать капли совершенно прозрачной жидкости, что свидетельствует о том, что перегоняется чистая вода.
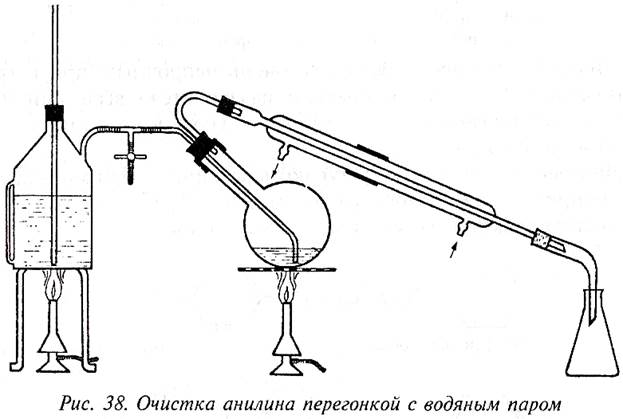
Дистиллят насыщают поваренной солью. Анилин нерастворим в насыщенном растворе хлорида натрия, поэтому жидкость расслаивается. Анилин отделяют с помощью делительной воронки, сушат небольшим количеством твердого гидроксида натрия или калия и перегоняют из небольшой колбочки с воздушным холодильником (темп. кип. 184°С).
Собирается довольно чистый продукт, в дальнейшем изменяющий окраску под действием кислорода воздуха. Полученного количества анилина вполне достаточно для постановки опытов с ним в нескольких классах.
§
С интересным и важным разделом органической химии — синтезом красителей — учащиеся могут быть ознакомлены лишь на внеклассных занятиях. Причем это знакомство по необходимости ограничивается простейшими представителями класса азокрасителей.
При синтезе красителей учащиеся знакомятся с двумя новыми реакциями — диазотированием и азосочетанием.
Здесь нецелесообразно излагать теорию красящих веществ, поэтому мы ограничимся лишь следующими указаниями.
При действии азотистой кислоты на соли первичных аминов
образуются так называемые диазосоединения. Соответствующая реакция называется реакцией диазотирования, например
C6H5NH2· HCI HNO2 à C6H5N2CI 2Н2O
гидрохлорид хлорид
анилина фенилдиазония
Диазосоединения — вещества очень непрочные: при нагревании выше 3— 5°С они, как правило, разлагаются с выделением азота; поэтому получают их обычно в растворах и реакцию диазотирования ведут при охлаждении.
Диазосоединения легко вступают в реакции с другими веществами, например с аминами и фенолами. Так образуются азокрасители, а сама реакция называется азосочетанием.
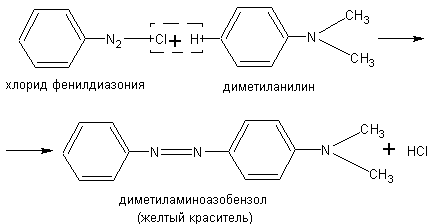
Синтез диметиламиноазобензола. Судя по приведенной структурной формуле желтого красителя, он должен получаться в результате азосочетания хлорида фенилдиазония C6H5N2CI с диметиланилином C6H5N(CH3)2:
Хлорид фенилдиазония, как указывалось выше, может быть получен действием азотистой кислоты на анилин. Практически вместо азотистой кислоты берут смесь соляной кислоты и нитрита натрия, при взаимодействии которых и образуется азотистая кислота.
Процесс выражается следующими уравнениями:
C6H5NH2 HCI à C6H5NH2 · HCI
NaNO2 HCI à HNO2 NaCI
C6H5NH2HCI HNO2 à C6H5N2CI H2O
или суммарно:
C6H5NH2 NaNO2 2HCI à C6H5N2CI NaCI 2H2O
Из уравнения следует, что для успешного протекания данной реакции на 1 моль анилина требуется 1 моль нитрита натрия и 2 моля HCI.
Для нашего опыта количество веществ можно уменьшить, например, в двадцать раз, т.е. можно взять 5 г анилина, 4 г соли и такое количество соляной кислоты, в котором содержится 3,6 г хлороводорода.
В химическом стакане смешивают взятое количество анилина с 50 мл соляной кислоты, содержащей 3,6 г НС1 (для этого кислоту, уд. вес 1,19, предварительно разбавляют тремя объемами воды). Добавляют в стакан несколько кусочков льда, а сам стакан ставят в воду со льдом. После того как полученный раствор охладится (температура его должна быть около 5°С), к нему при тщательном размешивании добавляют понемногу из капельной воронки указанное выше количество нитрита натрия в виде концентрированного водного раствора (предварительно также охлажденного). Нитрит натрия с избытком соляной кислоты дает азотистую кислоту, которая и диазотирует анилин. В растворе образуется хлорид фенилдиазония.
Полноту диазотирования анилина можно проверить иодокрах-мальной бумажкой: если анилин продиазотирован весь, то избыток азотистой кислоты выделит (окислит) иод из иодида калия и бумажка посинеет. Пробы производят через 2—3 мин после прибавления нитрита натрия. Если бумажка не синеет, добавляют еще немного раствора соли.
Для получения красителя к приготовленному раствору хлорида фенилдиазония приливают теоретическое количество диметилани-лина. Образуется краситель желтого цвета. Его отделяют от раствора и оформляют для коллекции.
Синтез гелиантина (метилоранжа). Чтобы азокрасители лучше растворялись в воде, их получают часто не из анилина, а из сульфаниловой кислоты
В первой стации синтеза данного красителя диазотируют сульфаниловую кислоту, а на второй стадии проводят азосочетание полученного вещества с диметиланилином. При этом образуется хорошо известный индикатор метилоранж
По аналогии с предыдущим нетрудно видеть, что сульфаниловая кислота, нитрит натрия и диметиланилин реагируют в эквимолекулярных отношениях. Если для опыта взять по 0,05 моля веществ и притом учесть, что сульфаниловая кислота кристаллизуется с двумя молекулами воды, то потребуется 10,5 г сульфаниловой кислоты, 4 г нитрита натрия и 6 г диметиланилина.
Навеску сульфаниловой кислоты растворяют в 25 мл 2 М раствора гидроксида натрия {2 г NaOH в 25 мл раствора). В этом же растворе затем растворяют навеску нитрита натрия. После этого раствор охлаждают льдом и приливают к 25 мл 2 М раствора соляной кислоты, охлаждаемого льдом (кроме наружного охлаждения кусочки льда могут быть помещены в раствор).
Рассчитанное количество диметиланилина растворяют в 5 мл 1 М соляной кислоты, охлаждают льдом и к охлажденному раствору приливают полученный выше раствор диазобензолсульфокислоты. Происходит образование красителя. Приливают еще раствор гидроксида натрия до сильнощелочной реакции. Из раствора выделяется натриевая соль красителя в виде оранжево-коричневых лепесткообразных кристалликов. Через несколько часов краситель отфильтровывают, промывают на воронке 25 мл воды, тщательно отжимают на фильтровальной бумаге и сушат в фарфоровой чашке на водяной бане.
Гелиантином можно окрасить образец шерсти или шелка. В химический стакан наливают раствор гелиантина, прибавляют несколько капель соляной кислоты, погружают в раствор окрашиваемый образец и кипятят жидкость, помешивая палочкой. Затем окрашиваемый материал промывают водой и сушат.
Гелиантин под именем метилоранжа известен как индикатор. В кислой среде он окрашивается в розовый цвет, в нейтральной и щелочной — в желтый. Изменение окраски легко заметить, если немного красителя растворить в подкисленной воде и затем нейтрализовать раствор щелочью.
АМИДЫ КИСЛОТ
Изучение амидов включено в школьную программу органической химии в целях ознакомления учащихся с важнейшим представителем этого класса соединений — карбамидом (мочевиной), на-
ходящим все более широкое применение в сельском хозяйстве (в качестве азотного удобрения, для скармливания жвачным животным) и в промышленном органическом синтезе (получение полимеров). Общие сведения о данном классе соединений ограничиваются почти исключительно рассмотрением строения амидов как аминопроизводных карбоновых кислот. Весь фактический экспериментальный материал темы относится полностью к карбамиду.
§
Гидролиз карбамида. При нагревании с растворами кислот или щелочей карбамид подвергается гидролизу. При этом образуются аммиак и углекислый газ:
а) В колбу с 30 мл воды помещают 5 г карбамида и приливают половинный объем концентрированного раствора гидроксида натрия. Смесь нагревают, а выделяющийся аммиак обнаруживают при помощи палочки, смоченной концентрированной соляной кислотой.
б) Растворяют 0,5 г карбамида в 2—3 мл воды (в пробирке), добавляют двойной объем прозрачной баритовой воды и смесь кипятят. Влажной лакмусовой бумажкой и по запаху обнаруживают выделяющийся аммиак. Наблюдают образование осадка карбоната бария в результате взаимодействия угольной кислоты со щелочью.
Взаимодействие карбамида с азотной кислотой. Несмотря на наличие аминогруппы в молекуле, амиды кислот, как правило, не проявляют основных свойств. В этом сказывается влияние карбонила на аминогруппу. В связи с тем что в молекуле карбамида содержатся две аминогруппы, он проявляет свойства очень слабого основания и по одной из аминогрупп образует соли с некоторыми кислотами, например с азотной:
В демонстрационную пробирку помещают 20 мл воды и 3—4 г карбамида. К раствору приливают вдвое меньший объем концентрированной азотной кислоты. Смесь взбалтывают и охлаждают холодной водой из-под крана. Выпадает осадок соли азотной кис-
лоты. Опыт можно проводить аналогично предыдущему, но с меньшими количествами реактивов (берут 1-2 мл раствора карбамида).
Взаимодействие карбамида с щавелевой кислотой. Наглядной является реакция карбамида с щавелевой кислотой, Поскольку щавелевая кислота двухосновная, а в карбамиде основные свойства проявляет одна аминогруппа, то кислота реагирует сразу с двумя молекулами карбамида:
Готовят насыщенные на холоду небольшие количества растворов карбамида и щавелевой кислоты. Сливают в пробирке или колбе равные объемы растворов. Вскоре образуется кристаллический осадок.
Образование биурета. Характерной реакцией карбамида является превращение его при нагревании в биурет:
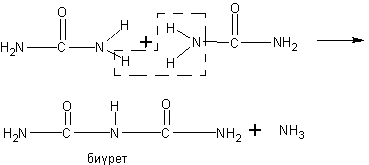
Нагревают в пробирке несколько кристалликов карбамида. Сначала он плавится, затем начинает разлагаться с выделением аммиака, который может быть обнаружен по запаху, при помощи стеклянной палочки, смоченной концентрированным раствором соляной кислоты, или другими способами. В пробирке образуется твердое белое вещество — биурет. Когда пробирка остынет, растворяют биурет в небольшом количестве воды при слабом нагревании. Затем добавляют немного раствора гидроксида натрия и 1-2 капли раствора сульфата меди. Образуется характерное для биурета (и
подобной группировки атомов в белках) красно-фиолетовое окрашивание.
§
Опыты с капроном. Капрон  рассматривается в курсе химии в связи с изучением аминокислот. Синтез капрона пока не разработан в качестве школьного опыта, хотя принципиально он вполне доступен. С капроном (в виде волокон, кусочков ткани, технических деталей) могут быть проведены следующие опыты.
рассматривается в курсе химии в связи с изучением аминокислот. Синтез капрона пока не разработан в качестве школьного опыта, хотя принципиально он вполне доступен. С капроном (в виде волокон, кусочков ткани, технических деталей) могут быть проведены следующие опыты.
а) В три пробирки с концентрированной серной кислотой, 10-процентным раствором щелочи и ацетоном помещают образцы капрона. Замечают, что растворение капрона происходит только в кислоте.
б) Нагревают капрон в пробирке на пламени горелки. Сначала он размягчается, затем плавится (216°С), при дальнейшем нагревании разлагается, выделяя летучие продукты с неприятным запахом.
в) Поджигают капроновое волокно. Оно горит с трудом, плавясь и образуя твердый, янтарного цвета шарик.
г) Кусочек капроновой смолы или свернутые в комочек обрывки трикотажного изделия нагревают в щипцах над пламенем горелки. Когда капрон начнет размягчаться и сделается вязким, прикасаются к нему лучинкой или стеклянной палочкой и, отводя затем ее быстро в сторону, вытягивают тонкие нити капрона, которые оказываются при этом довольно прочными.
Распознавание пластмасс. К окончанию курса органической химии у учащихся накопились уже достаточно сведений о пластмассах как теоретического, так и практического характера. Это позволяет поставить работы по распознаванию некоторых видов пластмасс.
Изделия из полиэтилена легче воды, жирные на ощупь, при нагревании быстро размягчаются, горят голубоватым пламенем, при этом ощущается слабый запах расплавленного парафина. В термостате при температуре 100—120°С они размягчаются. Для опыта могут быть использованы полиэтиленовая электроизоляция, пленочные материалы, флаконы, пробки от бутылок и т.д.
Полихлорвиниловые материалы распознаются по отщеплению хлороводорода при нагревании исследуемого образца в пробирке (обрезки клеенки, накидки, электроизоляции и т.п.). Хлороводо-род определяется по запаху, с помощью влажной лакмусовой бу-
мажки или по образованию дымка хлорида аммония при поднесении к пробирке палочки, смоченной раствором аммиака. Полихлорвинил горит в пламени с обильным выделением копоти. Размягчается при температуре около 60—66°С.
Изделия из политетрафторэтилена жирные на ощупь, матового цвета, напоминают по внешнему виду воск, не горят, плавятся лишь в сильном пламени.
Полистирол бывает прозрачный и непрозрачный, легко размягчается при нагревании, горит коптящим пламенем, выделяя при этом запах мономера. Изделия из полистирола хрупки, при постукивании звенят. Размягчается полистирол при 80°С. В качестве образцов для исследования могут быть использованы вазочки, розетки, обломки шкатулки, детские игрушки и т.п.
Полиметилметакрилат (листовое органическое стекло, письменные приборы, линейки, ручки, зубные щетки и т.д.) внешне очень похож на прозрачный полистирол. Отличить от последнего его можно по отсутствию звонкости и хрупкости, горит голубоватым пламенем без копоти, при этом слышится характерное потрескивание и выделяется эфирный запах.
Фенолформальдегидные пластмассы используются при изготовлении различных электродеталей черного и темно-коричневого цвета (штепсельные розетки, выключатели, телефонные трубки и другие изделия). При нагревании они не размягчаются и не плавятся, а разлагаются, при этом обнаруживается запах фенола и формальдегида. Фенол можно обнаружить, если порошкообразную пластмассу прокипятить в течение 20—30 мин с водой и затем на профильтрованный раствор подействовать бромной водой или раствором хлорида железа. Горят фенопласты только в пламени.
§
Несмотря на большое значение белков, школьный эксперимент по этой теме сравнительно беден. Он сводится обычно к опыту свертывания белка при нагревании и 1—2 цветным реакциям. В дополнение к этим работам следует считать весьма целесообразными опыты по обнаружению отдельных элементов в белках и опыты, иллюстрирующие коллоидное состояние белков в растворах.
Открытие к белках азота. Если понятие об определении углерода, водорода и галогенов было дано в первых темах курса органической химии, то здесь, прежде всего, следует ознакомить учащихся с открытием в белках азота и серы.
В пробирку помещают немного сваренного вкрутую или жидкого яичного белка, прибавляют двойное количество измельченной натронной извести и нагревают. В присутствии щелочи азот выделяется из белка в виде аммиака, который обнаруживают по запаху, по посинению поднесенной к отверстию пробирки влажной лакмусовой бумажки или при помощи стеклянной палочки, смоченной концентрированной соляной кислотой.
Открытие в белках серы. Сущность опыта состоит в том, что органическое вещество разрушают нагреванием со щелочью или сплавлением с металлическим натрием. Сера при этом выделяется в виде сульфида натрия Na2S, после чего се нетрудно открыть по образованию характерного осадка сульфида свинца PbS.
Осторожно нагревают в пробирке яичный белок или кусочек белой шерсти с концентрированным раствором щелочи до полного растворения белка. Образующийся сульфид натрия переходит в раствор. К полученному раствору приливают ацетат свинца. Выпадает черный осадок сульфида свинца.
Примечание. Открытие азота и серы можно провести в одном опыте. Белок кипятят с крепким раствором щелочи. Обнаруживают выделяющийся аммиак. После растворения белка разбавляют раствор водой и прибавляют раствор свинцовой соли.
Денатурация белков при нагревании.
а) Водный коллоидный раствор белка нагревают в химическом стаканчике. Образуется белый осадок (хлопья) свернувшегося белка. Белок — отфильтровывают и пытаются растворить в чистой воде. Растворение не происходит ни в обычных условиях, ни при нагревании. Учитель объясняет значение этих процессов в жизни организмов.
б) Нагревают немного молока в химическом стакане. Сверху образуется пленка свернувшегося белка. Ее поднимают стеклянной палочкой и демонстрируют. Затем пленку переносят в пробирку и оставляют для следующих опытов (для цветной реакции).
§
а) К раствору белка в пробирке приливают несколько миллилитров раствора щелочи и затем несколько капель слабого раствора сульфата меди. Жидкость окрашивается в фиолетовый или красно-фиолетовый цвет.
б) Немного пшеничной муки (чайную ложку) замешивают в фарфоровой чашке с водой. Образующееся тесто помещают в марлю и, разминая пальцами, промывают в стакане с водой до тех пор, пока из теста не будет стекать прозрачная жидкость. Таким способом отмывается из муки крахмал, в остатке содержится белок. Белок кипятят в пробирке с водой и затем проводят биуретовую реакцию.
С одними и теми же белками можно выполнить обе цветные реакции. Кипятят белую шерстяную ткань или волосы с раствором щелочи. Разливают раствор в две пробирки. В одной пробирке проводят биуретовую реакцию, в другой — ксантопротеиновую.
Этот опыт, кстати, поясняет, почему капли щелочи губительно действуют на шерстяную одежду и почему при стирке шерстяные изделия нельзя кипятить с содой.
Горение как способ распознавания белковых материалов. Вносят в пламя пучок шерсти, волос, перья птицы и другие белковые тела. Ощущается характерный запах жженых перьев или паленого.
Этот способ может быть использован как наиболее простой для распознавания белков. Например, чтобы отличить шерстяную пряжу от хлопчатобумажной или натуральный шелк от искусственного, можно поджечь очень небольшие пучки нитей исследуемых образцов.
Шерсть и натуральный шелк горят медленно и, как белковые тела, дают при этом характерный запах. Хлопковое волокно и искусственный шелк, представляющие собой в основном клетчатку, горят быстро, выделяя запах жженой бумаги.
ЛИТЕРАТУРА
1. Агроскин А.А., Панина Е.Ф. Лабораторные работы по химии и технологии угля. М.: Высшая школа, 1961.
2. Андрианов К. и Кардашев Д. Практические работы по искусственным смолам и пластмассам. М., 1946.
3. Афанасьев М.А., Агаханянц В.А., Королев Д.П., Тулякова Г.М. Количественные опыты по химии. М.: Просвещение, 1964.
4. Барков С.А., Курочкин А.А. Работы по органической химии. М., 1930.
5. Верховский В.Н., Гольдфарб Я.Л., Сморгонский Л.М. Методика преподавания химии в средней школе (раздел органической химии). М.: Учпедгиз, 1936.
6. Волжский И.А., Львов В.П., Рейсфельд В.О. Руководство к практическим занятиям в лаборатории синтетических каучуков. М., Госхимиздат, 1955.
7. Гаттерман А., Виланд Г. Практические работы по органической химии. М., 1948.
8. Голлеман А. Простые опыты по органической химии. М., 1939.
9. Гостев М.М. Химический кружок в школе. М.: Изд-во АПН РСФСР. М., 1958.
10. Грабецкий А.А. Опыты по химии. М.: Изд-во АПН РСФСР, 1957.
11. Девятова Н.И., Забоев С.А. Практикум по органической химии. М., 1941.
12. Егоркин В.Ф., Кирюшкин Д.М., Полосин B.C. Внеклассные практические занятия по химии. М.: Просвещение, 1965.
13. Збарский Б.И., Збарский И.Б., Солнцев А.И. Практикум по органической химии. М., 1949.
14. Зданчук Г.А. Химический кружок. М.: Просвещение, 1964.
15. Зонис С.А., Мазуров С.М. Лабораторно-лекционные опыты и демонстрационные материалы по органической химии. М.: Высшая школа, 1961.
16. Иванцова М.А., Левченко В.В. Практические работы по химии в школе с малым количеством реактивов. М., Учпедгиз, 1949.
17. Ключников Н.Г. Практические занятия по химической технологии. М.: Просвещение, 1965.
18. Кобазев И.А. Лабораторно-практические занятия по органической химии. М., 1958.
19. Копылев Б.А., Трабер А.Г., Сычев М.М., Григор В.А. Руководство к практическим занятиям по общей химической технологии. М.: Госхимиздат, 1963.
20. Крапивин С.Г. Практические работы по органической химии. М., 1910.
21. Крайнев С.И., Пятницкий Н.П. Практикум по органической химии. М., 1957.
22. Крешков А.П., Курбатов Н.И. Лабораторные работы по синтезу и анализу органических соединений. М., 1940′.
23. Лазурьевский Г.В., Терентьева И.В., Шамшурин А.А. Практические работы по химии природных соединений. М.: Высшая школа, 1961.
24. Лосев И.П., Федотова О.Я. Практикум по химии высокомолекулярных соединений. М.: Госхимиздат, 1959.
25. Лукашевич И.П., Смидович Е.В. Практикум по технологии нефти. М., 1947.
26. Малахов Б.П. Школьные работы по химической технологии органических веществ. М., 1928.
27. Парменов К.Я. Опыты по химии с электрическим током. Мл Учпедгиз, 1962.
28. Платковская В.М. Краткий курс практических занятий по органической химии. М., 1930.
29. Плетнер Ю.В., Полосин B.C. Практикум по методике обучения химии. М.: Учпедгиз, 1962.
30. Полосин B.C. Школьный эксперимент по неорганической химии. М.: Учпедгиз, 1959.
31. Прянишников Н.Д. Практикум по органической химии. М., 1949.
32. Рево А.Я. Качественные микрохимические реакции по органической химии. М.: Высшая школа, 1965.
33. Рупе Г. Лекционные опыты по органической химии / Пер. перераб. и доп. В.А.Измаильского. М., 1936.
34. Смирнов B.C. Практические работы по органической химии. М., 12935.
35. Смолин А.П., Рождественская В.А. Практические работы по органической и биологической химии. М.: Просвещение, 1964.
36. Стефановская З.Ф. Руководство к лабораторным занятиям по органической химии. М.: Высшая школа, 1964.
37. Чертков И.Н. Эксперимент по полимерам к средней школе. М.: Учпедгиз, 1961.
38. Шарпенак А.Э., Косенко С.А. Практикум по органической химии. М.: Высшая школа, 1965.
39. Щукина А.И. Руководство для проведения опытов по органической химии в средней школе. Куйбышевское издательство, 1962.
40. Юрьев Ю.К. Практические работы по органической химии. Вып. I, II, III. M.: Изд-во МГУ, 1964.
41. Stapf— Hradetzky. Chemische Schulfersuche, Teil 3, Berlin, 1962.
42. Gerhard Meyendorf, Einfache chemische Schulfersuche. Berlin, 1961.
* * *
43. Агрономов А.Е., Щабаров Ю.С. Лабораторные работы в органическом практикуме. М.: Химия, 1974.
44. Васильева Н.В., Куплетская Н.Б., Смолина Т.А. Практические работы по органической химии. М.: Просвещение, 1978.
45. Вейганд К., Хильгетаг Г. Методы эксперимента в органической химии / Пер. с нем. М.: Химия, 1968.
46. Назарова Т.С., Грабецкий А.А., Лаврова В.Н. Химический эксперимент в школе. М.: Просвещение, 1987.
47. Некрасов В.В. Руководство к малому практикуму по органической химии. М.: Химия, 1964, 1975.
48. Органикум. Практикум по органической химии: В 2 т. / Пер. с нем. М.: Мир, 1979.
49. Полосин B.C., Прокопенко В.Г. Практикум по методике преподавания химии. М.: Просвещение, 1989.
50. Степанов Б.И. Введение в химию и технологию органических красителей. М.: Химия, 1984.
51. Чертков И.Н., Жуков П.Н. Химический эксперимент с малыми количествами реактивов. М.’. Просвещение, 1989.
52. Шарп Дж., Госни И., Роули А. Практикум по органической химии / Пер. с англ. М.: Мир, 1993.
[1] Д.И.Менделеев. Основы химии. Изд. 4-е. 1881. С. V. (Предисловие)
[2] Там же. С. XII.
1 В растворе поваренной соли метан и хлор растворяются в меньшей степени, чем в чистой воде.
* практически пятиуксусный эфир получается действием на глюкозу не уксусной кислоты, а уксусного онгидрида
. 
Каталитическое жидкофазное окисление фенола кислородом воздуха в присутствии катализаторов высокодисперсного рутения, нанесенного на углеродные нанотрубки
Вестник Томского государственного университета. Химия. 2022. № 11. С. 65-82
УДК 544.472.2; 547.458.8 Б01 10.17223/24135542/11/6
А.Н. Субоч1′ 2, Н.В. Громов1′ 3, Т.Б. Медведева1, Л.С. Кибис1′ 2, Е.Ю. Герасимов1, А.Б. Аюшеев4, О.Ю. Подъячева1′ 2, О.П. Таран1, 3
1Институт катализа им. Г.К. Борескова СО РАН (г. Новосибирск, Россия) 2Новосибирский национальный исследовательский государственный университет
(г. Новосибирск, Россия) 3Новосибирский государственный технический университет (г. Новосибирск, Россия) 4 ООО «НИОСТ» (г. Томск, Россия)
Каталитическое жидкофазное окисление фенола кислородом воздуха в присутствии катализаторов высокодисперсного рутения, нанесенного на углеродные нанотрубки
Работа нацелена на создание катализаторов глубокого окисления органического загрязнителя фенола. Катализаторы — наночастицы рутения (1,4—1,9 нм, 3 вес. %), нанесенные на модифицированные азотом (0, 2, 3 и 6 вес. %) бамбу-коподобные углеродные нанотрубки. Катализаторы и их носители исследованы рядом физико-химических методов анализа (ПЭМ, адсорбция азота, РФЭС, АЭС-ИСП) и испытаны в превращении фенола при 160°С. Удалось добиться 100% конверсии фенола за два часа реакции.
Ключевые слова: органический загрязнитель; фенол; жидкофазное окисление; кислород; воздух; рутений; углеродные нанотрубки.
Введение
Многолетний экологический мониторинг, осуществляемый Министерством природных ресурсов и экологии Российской Федерации и находящейся в его ведении Федеральной службой по гидрометеорологии и мониторингу окружающей среды (Росгидромет), свидетельствует о достаточно напряженной экологической ситуации на водных объектах России. Такие данные приводятся в публикуемых в открытом доступе Росгидрометом ежегодных обзорах состояния и загрязнения окружающей среды в Российской Федерации [1]. Из-за негативного антропогенного воздействия десятки водных объектов России характеризуются крайне напряженной экологической обстановкой. Вода в этих объектах десятилетиями оценивается как «грязная», «очень грязная», а в отдельных районах как «экстремально грязная».
Среди экологически опасных веществ, попадающих в водоемы в результате хозяйственной деятельности человека, особое место занимают фенол и его производные — трудноокисляемые органические вещества, широко применяемые в химической, фармацевтической, кожевенной и других отраслях промышленности. Они попадают в окружающую среду преимущественно в составе промышленных сточных вод и могут оказы-
вать сильное токсикологическое действие на микроорганизмы, животных и человека [2, 3]. Фенол, а также его производные могут составлять до 25% загрязняющих веществ, попадающих в окружающую среду со сточными водами. Являясь простейшим представителем органических загрязнителей фенолового ряда, сам фенол часто используется как модельный компонент для исследований в области органической химии, катализа и экологии, направленных на создание новых или усовершенствование существующих технологий очистки сточных вод от фенольных загрязнителей.
Фенол и его производные не могут быть обезврежены традиционными биологическими методами очистки, применяемыми на муниципальных очистных сооружениях, ввиду их высокой токсичности для биоценозов микроорганизмов, используемых для извлечения органических веществ из стоков [4]. Альтернативный способ обезвреживания стоков от трудноокис-ляемых органических соединений, в том числе фенола, — жидкофазное окисление кислородом воздуха (Wet air oxidation). Такой метод нашел достаточно широкое применение в странах Западной Европы и США. Впервые жидкофазное окисление кислородом воздуха было запатентовано Ф. Зиммерманном примерно 60 лет назад [5] и апробировано в промышленности в конце 1950-х гг. Начиная с 1960-х гг. активно реализуются установки некаталитического высокотемпературного окисления фенолов и других органических соединений в сточных водах (температура от 240 до 280°С и давление воздуха 8,5-12 МПа) [6]. При этом предлагаемая технология предполагает протекание глубокого окисления фенолов с образованием экологически более безопасных продуктов — диоксида углерода и воды. Применение высоких температур и давлений требует существенных капитальных и текущих затрат при эксплуатации очистных установок. Повышение эффективности (в первую очередь за счет снижения температуры процесса и давления кислорода) жидкофазного окисления кислородом воздуха возможно путем разработки и применения катализаторов [7-10].
На сегодняшний день предложены как растворимые, так и твердые каталитические системы для удаления фенола и его производных путем их жидкофазного окисления кислородом воздуха. Гомогенные катализаторы, представленные солями и комплексами переходных металлов (например, Fe, Cu, Mn), достаточно эффективны в процессах окисления органических субстратов [11]. Вместе с тем растворимые каталитические системы обладают рядом недостатков, среди которых можно выделить попадание ионов металлов в сточные воды, трудности в регенерации катализаторов, а также их дезактивация за счет образования устойчивых комплексных соединений. Твердые катализаторы на основе высокодисперсных благородных металлов, например Ru, Pt, Pd, нанесенных на углеродные носители, представляются более перспективными системами для жидкофазного окисления органических загрязнителей. В отличие от растворимых катализаторов, твердые системы обладают рядом преимуществ, а именно высокой химической и механической стабильностью, низкой степенью вымывания активного компонента с углеродных носителей, легкостью отделения ката-
лизатора от реакционной среды и относительно простой регенерацией [12]. Причем именно Ru-содержащие катализаторы представляются наиболее перспективными в процессе окисления органических загрязнителей, в том числе фенола, по сравнению с другими катализаторами на основе металлов платиновой группы. Высокая активность систем Ru/С в жидкофазном окислении фенола была продемонстрирована авторами данной работы наших предыдущих исследованиях [13-15]. При создании эффективных катализаторов Ru/C большое внимание уделяется путям их модификации с целью повышения активности. Среди таких методов отдельно можно выделить внедрение азота в структуру углеродного носителя [16]. Допирование углеродных материалов азотом позволяет управлять электронными свойствами поверхности ряда углеродных материалов (в первую очередь углеродных нанотрубок, нановолокон) [16], а использование модифицированных азотом углеродных материалов дает возможность увеличить активность каталитических систем в реакциях окисления [17].
В заключение можно отметить, что в России не существует широко применяемых технологий для обезвреживания сточных вод от трудноокисляе-мых органических загрязнителей с помощью методов каталитического жид-кофазного окисления кислородом воздуха. Необходимость создания таких методов делает перспективными исследования, направленные на разработку фундаментальных основ указанных технологий. Ранее нами был исследован процесс жидкофазного окисления стойких органических загрязнителей на примере фенола в присутствии катализаторов на основе наночастиц рутения, нанесенных на модифицированные азотом углеродные нановолокна [18].
Целью настоящей работы стало продолжение систематических исследований по созданию эффективных катализаторов окисления фенола, а именно приготовление, физико-химические исследования и испытания в исследуемом процессе катализаторов высокодисперсного рутения, нанесенного на модифицированные азотом углеродные нанотрубки (Ru/N-МУНТ).
Экспериментальная часть
Приготовление носителей МУНТ. Углеродные нанотрубки, допирован-ные азотом, были синтезированы разложением этилен-аммиачной смеси на катализаторе 62Fe-8Ni-Al2O3 в проточном реакторе с виброожижением при 600-700°С [19]. Концентрацию аммиака в исходной смеси варьировали от 25 до 75 об. %. Для удаления адсорбированного кислорода с поверхности катализаторы подвергались предварительной восстановительной in situ обработке в течение 15 мин при температуре синтеза. Состав газовой смеси на выходе из реактора измеряли с помощью хроматографического анализа. Реакцию проводили до дезактивации катализатора. По окончании процесса реактор остужали в токе аргона. Объемную долю аммиака в этилен-аммиачной смеси и температуру синтеза варьировали для приготовления азотсодержащих МУНТов с заданным содержанием азота в структуре. Углеродные нанотрубки готовили по аналогичной методике без добавления аммиака [19].
Катализатор для приготовления МУНТов, содержащий 62 вес. % железа и 8 вес. % никеля (суммарное содержание металлов 70 вес. %) и 30 вес. % оксида алюминия, приготовлен методом соосаждения из водных растворов азотнокислых солей согласно [20, 21]. В качестве исходных реактивов были использованы 1,5 М растворы нитратов Ni(II), Fe(III) и Al(III), а в качестве осадителя — водные растворы NaOH, NH4HCO3 или NH4OH. Полученные осадки подвергали старению в маточном растворе при комнатной температуре в течение 1,5 ч, затем тщательно промывали, сушили на воздухе при комнатной температуре и затем при 110°С. Высушенные осадки прокаливали в токе воздуха при 450°С в течение 3 ч, а затем образцы восстанавливали в токе водорода при давлении 1 атм в проточном реакторе с виброожижен-ным слоем катализатора. После восстановления катализатор охлаждали в токе водорода, проводили пассивацию этанолом in situ и сушили на воздухе при комнатной температуре до сухого состояния образца. Время окончательной сушки катализатора достигало 48 ч.
Приготовление катализаторов окисления фенола. Высокодисперсные катализаторы окисления фенола на основе 3 вес. % Ru, нанесенного на МУНТ, готовили методом пропитки носителей по влагоемкости водным раствором предшественника металла нитрозилнитрата рутения Ru(NO)(NO3)3 с последующим восстановлением предшественника катализатора в токе водорода при 300 С (скорость нагрева 1 С/мин от комнатной температуры до 300°С) в течение 2 ч [18].
Физико-химические методы исследования катализаторов. Текстурные характеристики МУНТов и катализаторов Ru/МУНТ исследовали методом низкотемпературной адсорбции N2 при -204°C на установке ASAP-2400 (Micromeritics, США). Все образцы предварительно дегазировали в вакууме при 130-150°C.
РФЭС спектры МУНТ и Ru/МУНТ были записаны на фотоэлектронном спектрометре ES-300 (KRATOS Analytical) в режиме постоянной энергии пропускания энергоанализатора фотоэлектронов, который оснащен системой автоматизации на основе IBM PC. Для съемки использовался источник рентгеновского излучения без монохроматора. Энергия излучения AlKa составляла 1 486,6 эВ. Калибровка энергетической шкалы проводилась по энергии связи золота Au4f7/2, равной 84,0 эВ. Качественный контроль химического состава поверхности осуществлялся по обзорным спектрам в диапазоне 0 1 200 эВ с энергетическим разрешением, соответствующим максимуму чувствительности: энергия пропускания энергоанализатора (HV) — 50 эВ и шаг развертки — 1эВ. Для анализа количественного состава и химического состояния элементов проводилась съемка регионов внутренних уровней элементов (Cl2p, C1s, O1s, N1s, Fe2p3/2) и использовался режим: энергия пропускания энергоанализатора (HV) — 25 эВ, шаг развертки — 0,1эВ.
Элементный состав (содержание Ru и Fe) МУНТ и Ru/МУНТ исследовался методом атомно-эмиссионной спектроскопии с индуктивно-связанной плазмой (АЭС-ИСП).
Исследование размера высокодисперсных частиц рутения катализаторов Ru/МУНТ просвечивающей электронной микроскопией (ПЭМ) проводили на приборе JEM-2022 (JEOL, Япония) с ускоряющим напряжением 200 кВ и разрешением 1,4 А.
Испытание катализаторов проводили в автоклаве высокого давления (Autoclave Engineers, USA) при интенсивном перемешивании (1500 об./мин) [8], температуре и в окислительной атмосфере воздушной смеси, содержащей 20% О2 и 80% N2 [18]. Для выполнения экспериментов в реактор загружали 75 мл раствора фенола с начальной концентрацией субстрата 21 ммоль/л. Затем прибавляли 125 мг катализатора. Автоклав закрывали, продували аргоном 3 раза и искусственной воздушной смесью 3 раза. После промывки газами в реакторе устанавливали давление искусственной воздушной смеси 50 атм (парциальное давление кислорода 10 атм). Автоклав нагревали до температуры реакции 160°С. После достижения заданной температуры процесса (время нагревания составляло примерно 20-30 мин) отбирали нулевую пробу. В ходе реакции из автоклава периодически (0, 0,5, 1, 2, 3, 4, 5 и 6 ч) отбирали пробы реакционной смеси для анализа общего органического углерода (ООУ) и методом ВЭЖХ.
Аналитические методики. ВЭЖХ анализ содержания фенола в реакционной смеси проводили на хроматографе Милихром А-02 (ООО «ЭкоНОВА», г. Новосибирск, Россия), оборудованном спектрофотометрическим детектором. Анализ проводили на колонке Nucleosil C-18, термостатированной при 35°С. Элюент 20% ацетонитрила (0 сорт для ВЭЖХ хроматографии) и 80% 0,05 М ацетата аммония прокачивали через колонку со скоростью 150 мкл/мин. Время анализа составляло 11 мин. Анализ ООУ проводили на анализаторе углерода Analytik Jena Multi N/C 2100 S (Германия).
Глубина превращения фенола (конверсия субстрата) определялась как выраженное в процентах отношение разницы исходной и действительной концентраций фенола, отнесенной к начальному содержанию субстрата в реакционном растворе, согласно формуле:
^рюн(%) = СPhOH0″СPhOH • 100 , (1)
^PhOH
где Хаон — конверсия субстрата, %, С°юн и Срюн — начальная и действительная концентрации фенола, моль/л.
Изменения баланса общего органического углерода в реакционной смеси определялись в процентном выражении как отношение ООУ исследуемой пробы к исходной величине ООУ до реакции. Ниже приведена формула расчета баланса ООУ:
ООУ(%) = Сооу 7 Сооу -100, (2)
сооу
где ООУ — баланс общего органического углерода, %, Сюу и Сооу начальная и действительная концентрации фенола, мг/л.
Результаты и их обсуждение
Для проведения исследований по жидкофазному окислению кислородом воздуха модельного органического загрязнителя фенола приготовлены четыре углеродных носителя, представляющих собой модифицированные азотом нанотрубки. Содержание азота в образцах составляло 0, 2, 3 и 6 вес. % (табл. 1).
Таблица 1
Текстурные характеристики, размер наночастиц Ru и содержание элементов на поверхности носителей М-МУНТ и катализаторов Ru/N-МУНТ
Катализатор Текстурные характеристики катализаторов1 Размер Яи2 Содержание элементов3
Ае Уе Опор Уц <<!яи> С N Ее
м2/г см3/г нм см3/г нм вес. % вес. % вес. %
МУНТ 166 0,704 169 0 — 95 0 < 1
2%Ы-МУНТ 169 0,462 110 0 — 90 2 < 1
3%Ы-МУНТ 160 0,509 127 0 — 86 3 1
6%Ы-МУНТ 150 0,472 126 0 — 83 6 2
30/<^и/МУНТ 158 0,69 174 0 1,9 — — —
3%Яи/2%Ы-МУНТ 172 0,723 169 0 1,6 — — —
3%Яи/3%Ы-МУНТ 158 0,567 144 0 1,5 — — —
3%Яи/6%Ы-МУНТ 158 0,617 156 0 1,4 — — —
Примечания. 1. Ае — удельная поверхность; VI — суммарный объем пор; Vц — объем микропор; Dпор — диаметр пор.
2. Данные величины среднего размера наночастиц рутения приведены согласно результатам исследования поверхности методом ПЭМ.
3. Весовое содержание элементов на поверхности МУНТов приведено согласно результатам РФЭС до нанесения Ru.
На поверхность носителей наносился высокодисперсный металлический рутений в количестве 3 вес. %. Приготовленные носители Ы-МУНТ и катализаторы Яи/Ы-МУНТ были исследованы с помощью ряда физико-химических методов анализа (низкотемпературная адсорбция N2, ПЭМ, РФЭС, АЭС-ИСП). Текстурные характеристики всех образцов были исследованы методом низкотемпературной адсорбции азота (см. табл. 1). На рис. 1 приведены изотермы адсорбции-десорбции азота для исследованных носителей и катализаторов.
Все изотермы относятся к IV типу, присутствует петля гистерезиса в области р/ро 0,7-1,0. Анализ изотерм позволяет проследить динамику характеристических параметров текстуры (величины удельной поверхности, объема макро- и микропор, диаметр пор) в зависимости от условий и способа приготовления катализаторов (см. табл. 1). В целом можно отметить, что приготовленные в данной работе углеродные нанотрубки и катализаторы на их основе характеризуются достаточно развитой удельной поверхностью, величина которой находилась в диапазоне 138-169 м2/г.
0,0-
1°/Жи/6%^МУНТ
со
3% Ru/6% N -МУНТ
0,0
«* *
> »> АА *
с; . З/Жи/З/^-МУНТ
«Г 0,0к…….
С) I 3°/Жи/2%^МУНТ
п п ш_и_
Ю | б/^-МУНТ
о.0,0
О | З/^-МУНТ
0,0
0,2
0,4
0,6
0,8
1,0
Относительное давление (р/рд)
Рис. 1. Изотермы низкотемпературной адсорбции азота образцов №МУНТ и катализаторов ЯиМ-МУНТ
Суммарный объем пор каталитических систем составляет 0,5-0,7 см3/г, средний диаметр пор колеблется в диапазоне 110-200 нм. Микропоры отсутствуют. Введение азота в структуру углеродных нанотрубок приводит к уменьшению как величины удельной поверхности, так и объема и диаметра пор по сравнению с нанотрубками, немодифицированными азотом. Наблюдается обратная пропорциональность между содержанием азота в структуре носителя и величинами текстурных параметров. Нанесение наночастиц рутения на ^МУНТы не приводит к существенным изменениям удельной поверхности, в то время как величины среднего диаметра и объема пор меняется разнонаправлено.
Исследование ^МУНТов методом ПЭМ показало, что синтезированные носители представляют собой однородные по структуре бамбукопо-добные трубки с диаметром 13-16 нм и длиной до 1-2 мм, в отличие от образца нанотрубок, немодифицированных азотом, средний диаметр которых составляет 8 нм (рис. 2).
Сегменты углеродных нанотрубок состоят в среднем из 5-10 графено-вых слоев, причем толщина перегородок практически не отличается от толщины стенок трубок. Исследование Ru/МУНТ методом ПЭМ показало, что рутений находится в катализаторах в высокодисперсном состоянии в виде наночастиц с достаточно узким распределением частиц по размерам (1,41,9 нм) (см. табл. 1, рис. 2). Средний размер частиц Ru уменьшается с увеличением содержания азота в ^МУНТ. Наименьший средний размер наночастиц Яи, равный 1,4 нм, получен для катализатора 3%Ru/6%N-МУНТ.
Рис. 2. Микрофотографии катализаторов Ru/МУНТ (а) и Ru/N-МУНТ (б — 3%Яи/2%Ы-МУНТ, в — 3%Яи/3%Ы-МУНТ, г — 3%Яи/6%Ы-МУНТ)
Исследование носителей методом РФЭС показало, что спектры С^ полученных МУНТ и ^МУНТ имеют структуру, характерную для углеродных нанотрубок. Положение максимума пика в районе 284,4-284,9 эВ и наличие плотности в области 290-292 эВ указывают на sp2-гибридную структуру углерода (рис. 3).
Энергия связи, е/
Рис. 3. Спектры углерода С18 носителей №МУНТ
в
г
Небольшой сдвиг максимума спектра С^ в сторону больших энергий связи и уширение пика, наблюдаемые для азотсодержащих образцов К-УНТ, говорят о внедрении азота в структуру нанотрубок [22]. По данным РФЭС, количество азота в К-МУНТ варьирует от 2 до 6 вес. %. Азот в К-МУНТ находится в пиридиноподобном (~ 398,5 эВ), пиррольном (~ 399,5 эВ), графитоподобном (~ 401,0 эВ), окисленном (~ 402,5 эВ) и молекулярном (~ 405 эВ) состояниях [22, 23]. На рис. 4 приведен спектр азота N1s для образца МУНТ с разложением на индивидуальные компоненты.
394 336 398 400 402 404 406 40« 410 394 396 398 400 402 404 406 408 410 «>Л 390 398 400 407 4Ш 4« 410
Энергия связи, еЧ Энергия связи, еУ Энергия связи, еУ
Рис. 4. Спектры азота N18 с разложением для носителей №МУНТ
Соотношение форм азота меняется с увеличением азота К-УНТ: доля пиридинового азота увеличивается, в то время как доля пиррольного азота уменьшается, доля капсулированного и графитоподобного N2 остается без изменений (табл. 2).
Таблица 2
Вклад пиков азота (в %) в общую интенсивность спектра N1«*
Образец ^пиридин. ^пиррол. ^графит. №Ох N2 (капсул.) №Ох
2°/<№МУНТ 16,4 13,3 33,3 11,1 25,9 —
3°/<№МУНТ 18,0 9,5 31,4 12,4 26,9 1,8
6/№МУНТ 23,0 7,7 31,4 11,3 24,6 2,0
6°/<№МУНТ1 23,8 12,0 29,2 10,6 24,4 —
*Данные приведены для катализатора после испытаний в жидкофазном окислении фенола.
Анализ состояния рутения для Ru/МУНТ и Ru/N-МУНТ осложняется частичным наложением линии Ru3d на интенсивный спектр углерода C1s в области 280-287 эВ. Поэтому для более достоверной интерпретации состояния рутения был проведен анализ линии рутения Ru3pз/2. На рис. 5 приведены соответствующие спектры для Ru-содержащих образцов катализаторов.
Анализ данных РФЭС показал, что для Ru/МУНТ и Ru/N-МУНТ основным является состояние рутения с энергии связи порядка 462,6-462,9 эВ. Согласно литературным данным, это значение Есв является промежуточным между состоянием рутения Ru0 (461-462 эВ) [24, 25] и Ru4 (~ 463464 эВ) [24-26]. Возможно, происходит образование мелких частиц рутения, которые являются частично заряженными. Для образца Ru/МУНТ значение Есв(Ru3pз/2) более близко к энергии связи металлического рутения, что может быть связано с большим размером частиц.
462.9
462.9
КыЭр,
455
460
465
470
455
460
465
Энергия связи, эВ
470
Энергия связи, эВ
Рис. 5. Спектры Ru3p для Ru-содержащих катализаторов (1 — 3%Яи/МУНТ, 2 -3%Яи/2%К-МУНТ, 3 — 3%Яи/3%К-МУНТ, 4 — 3%Яи/6%К-МУНТ)
Методом РФЭС также подтверждено наличие в структуре МУНТов железа (см. табл. 1). Присутствие этого металла в составе углеродного материала объясняется тем, что синтез МУНТов проводился на катализаторе, в состав которого входит большое количество железа. Несмотря на то, что синтезированные МУНТ и К-МУНТ тщательно отмывались соляной кислотой на стадии подготовки к испытаниям, полностью удалить Fe из МУНТ не удалось. Содержание железа в МУНТах растет с увеличением количества азота. Согласно результатам РФЭС и АЭС-ИСП, в образцах МУНТ и 2%К-МУНТ содержание железа составляет менее одного процента (< 1 вес. %), а в 3%К-МУНТ и 6%К-МУНТ около 1 и 2 вес. % соответственно. По данным ПЭМ, остаточные частицы железа частично закапсулированы и находятся в углеродной оболочке.
Разработанные катализаторы и их носители испытаны в процессе жид-кофазного окисления фенола кислородом воздуха. Реакции проведены в оптимальных условиях, определенных в ходе выполнения исследований превращения органического субстрата в присутствии катализаторов на основе Яи, нанесенного на модифицированные азотом углеродные наново-локна [18]. Испытания проводили в автоклаве при 160°С и в атмосфере воздуха (давление 50 атм). Кинетические кривые расходования фенола и данные изменения баланса ООУ в течение времени приведены на рис. 6.
В холостом эксперименте наблюдается очень медленное окисление фенола. За 6 часов реакции израсходовано ~ 15% субстрата (см. рис. 6, а). Глубина окисления фенола также оказалась невысокой (согласно балансу ООУ, см. рис. 6, б). Жидкофазное окисление фенола также изучено в присутствии чистых носителей МУНТ без наночастиц Яи. В присутствии углеродных нанотрубок конверсия фенола составила 60-80%. Однако выявить влияние азота на каталитическую активность носителей и, как следствие, на активность Яи-содержащих катализаторов не представляется возможным из-за содержания в каталитической системе заметного количества примесей железа.
I- Холостой -Л- 2% М-МУНТ > МУНТ 3% *и/2% М-МУНТ
3% Ru/МУНТ
Врем я, ч
-V 3% М-МУНТ
6% М-МУНТ 3% Ru/3% М-МУНТ 3% Ru/6% М-МУНТ 1% *и/6% М-МУНТ
ео
о о
20
„ о
Время, ч
^ 2% М-МУНТ 3% М-МУНТ 6% М-МУНТ
> МУНТ -А- 3% 1*11/2% М-МУНТ 3% *и/3% М-МУНТ 3% *и/6% М-МУНТ
3% *и/МУНТ 1% *и/6% М-МУНТ
а б
Рис. 6. Кинетические кривые окисления фенола кислородом воздуха в присутствии углеродных катализаторов (а — конверсия фенола, б — баланс ООУ).
Условия реакции: 0,021 М PhOH, 1,67 г/л катализатора, 160°С, Рвозд = 50 атм
Ранее нами было показано, что железо даже в незначительных количествах способно окислять органические субстраты [27]. В данной работе в составе МУНТов содержание железа достигает 2 вес. % и оказывает намного более сильное влияние на их активность по сравнению с азотом. В пользу сильного влияния железа на активность катализаторов свидетельствуют и кинетические кривые конверсии фенола. В присутствии МУНТ и 2%К-МУНТ, в которых содержание железа составляет менее 1%, кинетические кривые расходования фенола имеют индукционный период, связанный с замедленным вымыванием в раствор небольших количеств сильно кап-сулированного в структуре нанотрубок железа. В присутствии 3%К-МУНТ и 6%К-МУНТ, в которых содержание Fe достигает 1 и 2 вес. % соответственно, индукционный период не наблюдается, причем катализатор с большим содержанием железа проявляет заметно большую каталитическую активность. Вымывание железа в реакционную среду подтверждено анализом АЭС-ИСП. Таким образом, для выявления влияния азота на активность катализаторов наночастиц рутения, нанесенного на модифицированные азотом углеродные нанотрубки, необходимо проведение дополнительных исследований, направленных в том числе на удаление примесей железа из состава катализатора. Однако результаты исследований катализаторов ПЭМ позволяют отметить положительную роль азота, встроенного в структуру нанотрубок, на стабилизацию высокодисперсного состояния металла и заметное уменьшение среднего размера наночастиц Ru (с 1,9 нм для образца, немодифицированного азотом, до 1,4 нм на поверхности 6%К-МУНТ).
Анализ методом ВЭЖХ реакционных смесей, полученных в присутствии как Ru/МУНТ, так и Ru/N-МУНТ катализаторов, выявил уменьшение концентрации фенола уже в нулевой точке (70-80%), а в течение 30 мин концентрация фенола снижалась до 27-48%. Через 3 ч конверсия фенола составляет уже более 95% и достигает практически 100% по окончании экспериментов. Вместе с тем баланс ООУ после 6 ч составляет 25-30%, свидетельствуя о неполном протекании глубокого окисления органического субстрата и интермедиатов реакции в диоксид углерода и воду.
100
100
80
80
о 60 в-
40
40
Ч 20
4
5
В целом следует отметить, что активности 3%Ru-содержащих катализаторов довольно близки друг другу, а каталитическая система 3%Ru/6%N-МУНТ оказалась несколько активнее других Ru-содержащих образцов. Проведено исследование катализаторов 3%Ru/N-МУНТ и носителей ^МУНТ после реакции окисления фенола кислородом воздуха методами ПЭМ. Согласно результатам ПЭМ, изменений среднего размера наночастиц рутения не происходит. Изменение состояния рутения в ходе окисления фенола изучено методом РФЭС на примере пары наиболее активного катализатора 3%Яи/6%К-МУНТ и его носителя без благородного металла 6%^МУНТ. Установлено, что зарядовое состояние рутения в ходе реакции для всех образцов также практически не изменяется. Исследование состояния азота в 3%Ru/6%N-МУНТ и 6%^МУНТ после реакции показало небольшое увеличение доли пиррольного азота (см. табл. 2), а также уменьшение количества азота в катализаторе 3%Ru/6%N-МУНТ. Стоит отметить, что снижается и количество железа в катализаторе, вымывание которого дополнительно подтверждено исследованиями реакционных растворов методом анализа АЭС-ИСП.
При разработке новой каталитической системы очень важным параметром является стабильность катализатора (сохранение каталитической активности) в реакционных условиях в нескольких циклах процесса. Цикловые испытания в данной работе были проведены в присутствии наиболее активного катализатора 3%Ru/6%N-МУНТ. После первого цикла реакции катализатор был отделен от реакционной смеси фильтрацией, отмыт несколько раз деионизованной водой для удаления компонентов реакционной смеси и высушен. Этот образец был повторно испытан в реакции окисления фенола (рис. 7).
к
О 40 О
2 3 4 5
Время, ч
а
2 3 4 5
Время, ч
б
Рис. 7. Испытания катализатора 3%Ки/6%№МУНТ в жидкофазном окислении фенола в двух циклах (а — конверсия фенола, б — баланс ООУ). Условия реакции: 0,021 М PhOH, 1,67 г/л катализатора, 160°С, Рвозд = 50 атм
Оказалось, что активность 3%Ru/6%N-МУНТ снижается при повторном испытании. Так, через 1 ч концентрация фенола во цикле II в пять раз выше по сравнению с циклом I. Конверсия ООУ во цикле II ниже на 10-20%, чем в цикле I.
100
80
80
60
X 60
40
Ш 20
20
0
6
Выводы
В работе проведено исследование процесса каталитического жидкофаз-ного окисления органических загрязнителей кислородом воздуха на примере загрязнителя фенола. В качестве катализаторов исследовались системы наночастиц благородного металла рутения, нанесенные на поверхность модифицированных азотом углеродных нанотрубок (Ru/N-МУНТ). Также изучена каталитическая активность носителей без наночастиц металла. Изучено 4 образца катализаторов, содержащих по 3 вес. % Ru и отличающихся содержанием азота в структуре нанотрубок (0, 2, 3 и 6 вес. %). Испытания катализаторов проведены в гидротермальных условиях при 160°С и давлении воздушной смеси 50 атм. В результате проведенных исследований удалось установить, что катализаторы 3%Ru/N-MyHT высокоэффективны в окислении фенола. Удалось достичь 100%-ной конверсии субстрата и снижения баланса ООУ до 25-30%. Не содержащие Ru носители также проявили умеренную активность в исследуемом процессе. Однако из-за примесей в образцах нанотрубок железа (до 2 вес. %), оставшегося после отмывки носителя кислотами от Fe-содержащего катализатора синтеза углеродных трубок, выявить явным образом влияние азота на активность катализаторов не представлялось возможным. Вместе с тем зафиксировано уменьшение среднего размера наночастиц Ru на поверхности носителей с ростом содержания азота в структуре катализатора.
В целом можно отметить, что среди созданных катализаторов наибольшую активность в реакции жидкофазного глубокого окисления фенола продемонстрировал катализатор 3%Ru/6%N-MyHT, содержащий 6 вес. % азота. В присутствии каталитической системы наиболее перспективного состава концентрация фенола снижается до 1% уже через 3 часа реакции. Однако катализатор 3%Ru/6%N-MyHT, испытанный в двух циклах превращений фенола, продемонстрировал снижение активности при переходе ко второму циклу испытаний. Необходимо проведение дополнительных исследований в данной области, направленных на улучшение состава катализатора (удаление примесей железа) и повышение стабильности каталитических систем при их многоцикловых испытаниях.
Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (проект № 16-33-00631).
Литература
1. Обзор состояния и загрязнения окружающей среды в Российской Федерации за
2022 год / Федеральная служба по гидрометеорологии и мониторингу окружающей среды (Росгидромет) ; под ред. Г.М. Черногаевой и др. М., 2022. URL: http://www.meteorf.ru/product/infomaterials/90/
2. Новый справочник химика и технолога. Сырье и продукты промышленности
органических и неорганических веществ / ред. Ю.В. Поконова; В.И.Стархова. СПб. : Мир и Семья, Профессионал, 2005. Ч. I, II.
3. Филатов В.А., Иван Б.А., Мусийчук Ю.И. Кислородсодержащие органические
соединения. Ч. I // Вредные вещества в окружающей среде. СПб. : Профессионал, 2007. С. 107-113.
4. Autenrieth R.L., Bonner J.S., Akgerman A., Okaygun M. Biodegradation of phenolic
wastes // J. Hazard. Mater. 1991. V. 28, № 1-2. P. 29-53.
5. Пат. 2665249 США, МПК CI 210-2. Waste Disposal / F.J. Zimmermann, W.Wis;
заявитель и патентообладатель Sterling Drug Inc. Опубл. 5 янв. 1954.
6. Siemens : [официальный сайт компании]. URL: https://www.siemens.com/global/en/
home/markets/water/waste-water-treatment.html#
7. Cao Y., Li B., Zhong G. et al. Catalytic wet air oxidation of phenol over carbon nanotubes:
Synergistic effect of carboxyl groups and edge carbons // Carbon. 2022. V. 133. P. 464-473.
8. Gallezot P., Chaumet S., Perrard A. et al. Catalytic Wet Air Oxidation of Acetic Acid
on Carbon-Supported Ruthenium Catalysts // Journal of Catalysis. 1997. V. 168. № 1. P. 104-109.
9. Mishra V.S., Mahajani V.V., Joshi J.B. Wet air oxidation // Ind. Eng. Chem. Res. 1995.
V. 34, № 1. P. 2-48.
10. Zhao X., Ma J., Jiang J. et al. Phenols and anilines degradation by permanganate in the absence / presence of carbon nanotubes: Oxidation and dehalogenation // Separation and Purification Technology. 2022. V. 170. P. 344-352.
11. Luck F. Wet air oxidation: past, present and future // Catalysis Today. 1999. V. 53, № 1. P. 81-91.
12. Stuber F., Font J., Fortuny A. et al. Carbon materials and catalytic wet air oxidation of organic pollutants in wastewater // Topics in Catalysis. 2005. V. 33, № 1-4. P. 3-50.
13. Taran O.P., Descorme C., Polyanskayа E.M. et al. Sibunit Based Catalytic Materials for the Deep Oxidation of Organic Ecotoxicants in Aqueous Solutions. III: Wet Air Oxidation of Phenol over Oxidized Carbon and Ru/C Catalysts // Catalysis in Industry. 2022. V. 5, № 2. P. 164-174.
14. Taran O.P., Polyanskaya E.M., Ogorodnikova O.L. et al. Sibunit-based catalytic materials for the deep oxidation of organic ecotoxicants in aqueous solutions. II: Wet peroxide oxidation over oxidized carbon catalysts // Catalysis in Industry. 2022. V. 3, № 2. P. 161-169.
15. Taran O.P., Polyanskaya E.M., Ogorodnikova O.L. et al. Sibunit-based catalytic materials for the deep oxidation of organic ecotoxicants in aqueous solution: I. Surface properties of the oxidized sibunit samples // Catalysis in Industry. 2022. V. 2, № 4. P. 381-386.
16. Ismagilov Z., Shalagina A., Podyacheva O. et al. Structure and electrical conductivity of nitrogen-doped carbon nanofibers // Carbon. V. 47, № 8. P. 1922-1929.
17. Podyacheva O., Ismagilov Z., Boronin A. et al. Platinum nanoparticles supported on nitrogen-containing carbon nanofibers // Catalysis Today. 2022. V. 186, № 1. P. 42-47.
18. Ayusheev A., Taran O., Seryak I. et al. Ruthenium nanoparticles supported on nitrogen-doped carbon nanofibers for the catalytic wet air oxidation of phenol // Applied Catalysis B: Environmental. 2022. V. 146. P. 177-185.
19. Субоч А.Н., Кибис Л.С., Стонкус О.А. и др. Синтез и исследование многостенных углеродных нанотрубок, допированных азотом // Химия в интересах устойчивого развития. 2022. № 1. С. 85-91.
20. Reshetenko T.V., Avdeeva L.B., Khassin A.A. et al. Coprecipitated iron-containing catalysts (Fe-AkO3, Fe-Co-AkO3, Fe-Ni-AkO3) for methane decomposition at moderate temperatures. I. Genesis of calcined and reduced catalysts // Applied Catalysis A. 2004. V. 268. P. 127-138.
21. Avdeeva L.B., Kochubey D.I., Shaikhutdinov S.K. Cobalt catalysts of methane decomposition: accumulation of the filamentous carbon // Applied Catalysis A. 1999. V. 177. P. 43-51.
22. Susi T., Pichler T., Ayala P. X-ray photoelectron spectroscopy of graphitic carbon nanomaterials doped with heteroatoms // J. Nanotechnol. 2022. V. б, № 1. P. 177-192.
23. Bulusheva L.G., Okotrub A.V., Fedoseeva Y.V. et al. Controlling pyridinic, pyrrolic, graphitic, and molecular nitrogen in multi-wall carbon nanotubes using precursors with different N/C ratios in aerosol assisted chemical vapor deposition // Phys. Chem. Chem. Phys. 2022. V. 17, № 37. С. 23741-23747.
24. Zhang X., Chan K.-Y. Water-in-Oil Microemulsion Synthesis of Platinum-Ruthenium Nanoparticles, Their Characterization and Electrocatalytic Properties // Chem. Mater. 2003. V. 15, № 2. С. 451-459.
25. Cattaneo C., Sanchez de Pinto M.I., Mishima H. et al. Characterization of platinum-rutheniun electrodeposits using XRD, AES and XPS analysis // J. Elecroanal. Chem. 1999. V. 461. № 1-2. Р. 32-39.
26. Hsieh T.-F., Chuang C.-C., Chen W.-J. et al. Hydrous ruthenium dioxide / multi-walled carbon-nanotube / titanium electrodes for supercapacitors // Carbon. 2022. V. 50, № 5. P. 1740-1747.
27. Полянская Е.М. Исследование катализаторов на основе наноразмерных углеродных материалов в реакциях глубокого жидкофазного окисления органических субстратов кислородом и пероксидом водорода : дис. … канд. хим. наук : 02.00.15. Новосибирск, 2022. 161 с.
Информация об авторах:
Субоч Арина Николаевна, младший научный сотрудник лаборатории экологического катализа Института катализа им. Г.К. Борескова Сибирского отделения Российской академии паук (г. Новосибирск, Россия). E-mail: arina@catalysis.ru
Громов Николай Владимирович, канд. хим. наук, научный сотрудник лаборатории каталитических методов переработки солнечной энергии Института катализа им. Г.К. Борескова Сибирского отделения Российской академии наук (г. Новосибирск, Россия); доцент кафедры инженерных проблем экологии Новосибирского государственного технического университета (г. Новосибирск, Россия). E-mail: gromov@catalysis.ru
Медведева Татьяна Борисовна, инженер лаборатории каталитических методов переработки солнечной энергии Института катализа им. Г.К. Борескова Сибирского отделения Российской академии наук (г. Новосибирск, Россия). E-mail: tanmedvedeva@catalysis.ru
Кибис Лидия Сергеевна, канд. хим. наук, научный сотрудник группы исследования нанесенных металл-оксидных катализаторов Института катализа им. Г.К. Борескова Сибирского отделения Российской академии наук (г. Новосибирск, Россия); научный сотрудник лаборатории синтеза и физико-химических исследований новых композитных катализаторов Новосибирского националь-пого исследовательского государственного университета. E-mail: kibis@catalysis.ru Герасимов Евгений Юрьевич, капд. физ.-мат. паук, научный сотрудник лаборатории структурных методов исследования Института катализа им. Г.К. Борескова Сибирского отделения Российской академии паук (г. Новосибирск, Россия). E-mail: gerasimov@catalysis.ru
Аюшеев Артемий Буладович, канд. хим. наук, главный эксперт отдела управления проектами дирекции по новым продуктам и технологиям OOO «НИOСT» (г. Томск, Россия). E-mail: a.ayusheev@gmail.com
Подъячева Ольга Юрьевна, д-р хим. наук, старший научный сотрудник лаборатории экологического катализа Института катализа им. Г.К. Борескова Сибирского отделения Российской академии паук (г. Новосибирск, Россия); старший научный сотрудник лаборатории синтеза и физико-химических исследований новых композитных катализаторов Новосибирского национального исследовательского государственного университета. E-mail: pod@catalysis.ru Таран Оксана Павловна, д-р хим. наук, ведущий научный сотрудник лаборатории каталитических методов переработки солнечной энергии Института катализа им. Г.К. Борескова Сибирского отделения Российской академии наук (г. Новосибирск, Россия); профессор кафедры инженерных проблем экологии Новосибирского государственного технического университета (г. Новосибирск, Россия). E-mail: oxanap@catalysis.ru
Tomsk State University Journal of Chemistry, 2022, 11, 65-82. DOI: 10.17223/2413 5542/11 /6
A.N. Suboch1- 2, N.V. Gromov1- 3, T.B. Medvedeva1, L.S. Kibis1- 2, E.Yu. Gerasimov1, A.B. Ayusheev4, O.Yu. Podyacheva1′ 2, O.P. Taran1′ 3
1 Boreskov Institute of Catalysis Siberian Branch of the Russian Academy of Sciences (Novosibirsk, Russia) 2Novosibirsk National Research State University (Novosibirsk, Russia) 3Novosibirsk State Technical University (Novosibirsk, Russia) 4 NIOSTLtd. (Tomsk, Russia)
Catalytic wet air oxidation of phenol in the presence of catalysts based on highly dispersed Ru supported on carbon nanotubes
The large volumes of organic pollutants formed by various industrial plants are very dangerous for the environment and human health. Phenol and its derivatives are particularly harmful substances. An important problem in the field of phenol utilization is the impossibility of PhOH conversion by traditional biotechnologies due to phenol’s high toxicity with respect to microorganisms. This makes necessary to develop a new, efficient technology for processing ofphenol pollutants. Catalytic wet air oxidation is proposed as a method of utilization ofphenols. Both soluble (based on salts and complexes of transition metals) and solid (for example, highly dispersed platinum group metals supported on inert carbon substrates) catalysts are proposed for wet air oxidation of organic substrates, but solid catalytic systems are more promising than soluble ones due to easy regeneration and separation from the reaction medium.
The purpose of this work was the preparation of catalysts based on ruthenium na-noparticles supported on carbon nanotubes modified by nitrogen and testing of the catalysts prepared in the wet air oxidation of organic pollutants. The work is a logical development of our research aimed at creating methods for the environmentally friendly disposal of organic pollutants in the presence of highly dispersed ruthenium catalysts deposited on various types of carbon carriers (graphite-like material, sibunite, and carbon nanofibers).
Bamboo-like carbon nanotubes doped with nitrogen (N-MCNT) with a modifying additive content of 0, 2, 3 and 6 wt. % were synthesized. N-MCNT supports were prepared by catalytic decomposition of the ethylene-ammonia mixture in the presence of the 62Fe-8Ni-30AhO3 catalyst, and 3 wt. % of Ru nanoparticles were precipitated onto the prepared supports. The catalysts of 3%Ru/N-MCNT and their supports were investigated by a number of physico-chemical methods, namely TEM, nitrogen adsorption, XPS, and ACP-ICP. The composition of the catalysts was confirmed, the size of the Ru nanoparticles was found to be 1.4—1.9 nm, and no change in the structure of N-MCNTs occurred during the metal deposition. Precipitation of Ru did not lead to a significant change in the textural characteristics of the supports. The size of the metal nanoparticles was inversely proportional to the nitrogen content, i.e., nitrogen promoted the stabilization of small nanoparticles. An XPS study showed that 3% of Ru/N-MCNTs also contains atoms of Fe (up to 2 wt. %), which are the remains of the 62Fe-8Ni-30AhO3 catalyst that was not removed during washing. Developed 3 %Ru /N-MCNT catalysts and their N-MCNT supports were tested in the wet air oxydation of phenol used as an example organic pollutant at 160°C. Three percent Ru/N-MCNT catalysts was found to be highly effective in the utilization of phenol. In the presence of the catalytic systems created, 100% conversion ofphenol was achieved, with a residual balance of total organic carbon of 25-30% of the initial value. At the same time, the presence of iron impurities made it impossible to clearly show the impact of nitrogen modification of nanotubes on the catalytic activity of N-MCNT supports and,
consequently, on the activity of 3% Ru/N-MCNT catalysts. Further efforts in this field of research will be concentrated, among other things, on improving the quality of the catalyst composition.
Key words: organic waste; phenol; wet air oxidation; ruthenium; carbon nanotube.
References
1. Review of Condition and Pollution of the Environment of the Russian Federation in 2022.
Moscow, 2022. URL: http://www.meteorf.ru/product/infomaterials/90/. (In Russian)
2. New Hand-book for chemist and technologist. Raw materials and products of organic and
inorganic synthesis Industries. Part I, II. Ed. Yu.V. Pokonova; V.I. Starhova. Saint-Petersburg : World and Family, Professional, 2005. (In Russian)
3. Filatov V.A., Ivin B.A., Musiychuk Yu.I. Oxygen-containing organic compounds. Part I.
Harmful substances in the environment. Saint-Petersburg: Professional, 2007. P. 107-113. (In Russian)
4. Autenrieth R.L., Bonner J.S., Akgerman A., Okaygun M. Biodegradation of phenolic
wastes. J. Hazard. Mater. 1991;28(1-2):29-53.
5. Pat. 2665249 SShA. MPK CI 210-2. Waste Disposal / F.J. Zimmermann. W. Wis;
zayavitel i patentoobladatel Sterling Drug Inc. Opubl. 5 yanv. 1954.
6. Siemens. URL: https://www.siemens.com/global/en/home/markets/water/waste-water-
treatment.html#.
7. Cao Y., Li B., Zhong G. et al. Catalytic wet air oxidation of phenol over carbon nanotubes:
Synergistic effect of carboxyl groups and edge carbons. Carbon. 2022;133:464-473.
8. Gallezot P., Chaumet S., Perrard A. et al. Catalytic Wet Air Oxidation of Acetic Acid on
Carbon-Supported Ruthenium Catalysts. Journal of Catalysis. 1997;168(1):104-109.
9. Mishra V.S., Mahajani V.V., Joshi J.B. Wet air oxidation. Ind. Eng. Chem. Res.
1995;34(1):2-48.
10. Zhao X., Ma J., Jiang J. et al. Phenols and anilines degradation by permanganate in the absence / presence of carbon nanotubes: Oxidation and dehalogenation. Separation and Purification Technology. 2022;170:344-352.
11. Luck F. Wet air oxidation: past, present and future. Catalysis Today. 1999;53(1):81-91.
12. Stuber F., Font J., Fortuny A. et al. Carbon materials and catalytic wet air oxidation of organic pollutants in wastewater. Topics in Catalysis. 2005;33(1-4):3-50.
13. Taran O.P., Descorme C., Polyanskaya E.M. et al. Sibunit Based Catalytic Materials for the Deep Oxidation of Organic Ecotoxicants in Aqueous Solutions. III: Wet Air Oxidation of Phenol over Oxidized Carbon and Ru/C Catalysts. Catalysis in Industry. 2022;5(2):164-174.
14. Taran O.P., Polyanskaya E.M., Ogorodnikova O.L. et al. Sibunit-based catalytic materials for the deep oxidation of organic ecotoxicants in aqueous solutions. II: Wet peroxide oxidation over oxidized carbon catalysts. Catalysis in Industry. 2022;3(2): 161-169.
15. Taran O.P., Polyanskaya E.M., Ogorodnikova O.L. et al. Sibunit-based catalytic materials for the deep oxidation of organic ecotoxicants in aqueous solution: I. Surface properties of the oxidized sibunit samples. Catalysis in Industry. 2022;2(4):381-386.
16. Ismagilov Z., Shalagina A., Podyacheva O. et al. Structure and electrical conductivity of nitrogen-doped carbon nanofibers. Carbon. 2009;47(8):1922-1929.
17. Podyacheva O., Ismagilov Z., Boronin A. et al. Platinum nanoparticles supported on nitrogen-containing carbon nanofibers. Catalysis Today. 2022;186(1):42-47.
18. Ayusheev A., Taran O., Seryak I. et al. Ruthenium nanoparticles supported on nitrogen-doped carbon nanofibers for the catalytic wet air oxidation of phenol. Applied Catalysis B: Environmental. 2022;146:177-185.
19. Suboch A.N., Kibis L.S., Stonkus O.A. et al. Synthesis and investigation of multi-wall nitrogen-added carbon nanotubes. Khimia v interesah ustoichivogo razvitiya. 2022;1:85-91. (In Russian)
20. Reshetenko T.V., Avdeeva L.B., Khassin A.A. et al. Coprecipitated iron-containing catalysts (Fe-AkO3, Fe-Co-AkO3, Fe-Ni-Al2O3) for methane decomposition at moderate temperatures. I. Genesis of calcined and reduced catalysts. Applied Catalysis A. 2004;268:127-138.
21. Avdeeva L.B., Kochubey, D.I., Shaikhutdinov S.K. Cobalt catalysts of methane decomposition: accumulation of the filamentous carbon. Applied Catalysis A. 1999;177:43-51.
22. Susi T., Pichler T., Ayala P. X-ray photoelectron spectroscopy of graphitic carbon nanomaterials doped with heteroatoms. J. Nanotechnol. 2022;6(1):177-192.
23. Bulusheva L.G., Okotrub A.V., Fedoseeva Y.V. et al. Controlling pyridinic, pyrrolic, graphitic, and molecular nitrogen in multi-wall carbon nanotubes using precursors with different N/C ratios in aerosol assisted chemical vapor deposition. Phys. Chem. Chem. Phys. 2022;17(37):23741-23747.
24. Zhang X., Chan K.-Y. Water-in-Oil Microemulsion Synthesis of Platinum-Ruthenium Nanoparticles, Their Characterization and Electrocatalytic Properties. Chem. Mater. 2003;15(2):451-459.
25. Cattaneo C., Sanchez de Pinto M.I., Mishima H. et al. Characterization of platinum-rutheniun electrodeposits using XRD, AES and XPS analysis. J. Elecroanal. Chem. 1999;461(1-2):32-39.
26. Hsieh T.-F., Chuang C.-C., Chen W.-J. et al. Hydrous ruthenium dioxide / multi-walled carbon-nanotube / titanium electrodes for supercapacitors. Carbon. 2022;50(5): 1740-1747.
27. Polyanskaya E.M. Issledovaniye katalizatorov na osnove nanorazmernykh uglerodnykh materialov v reaktsiyakh glubokogo zhidkofaznogo okisleniya organicheskikh substratov kislorodom i peroksidom vodoroda : dis. … kand. khim. nauk : 02.00.15. Novosibirsk, 2022. 161 p. (In Russian)
Information about the authors:
Suboch Arina N., PhD student, Junior researcher in the Laboratory of Ecological Catalysis of the Boreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences (Novosibirsk, Russian Federation). E-mail: arina@catalysis.ru
Gromov Nikolai V., PhD, researcher at the Laboratory of Catalytic Methods of Solar Energy Transformation of the Boreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences (Novosibirsk, Russian Federation), associate professor, Department of Engineering Issues of Ecology, Novosibirsk State Technical University (Novosibirsk, Russia). E-mail: gromov@catalysis.ru Medvedeva Tatyana B., engineer of the Laboratory of Catalytic Methods of Solar Energy Transformation of the Boreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences (Novosibirsk, Russian Federation). E-mail: tanmedvedeva@catalysis.ru
Kibis Lidia S., PhD, researcher at the Group of Investigations of supported metal-oxide catalysts of the Boreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences (Novosibirsk, Russian Federation), researcher in the Laboratory of Synthesis and Physic-Chemical Investigations of New Composite Materials in the Novosibirsk National Research State University. E-mail: kibis@catalysis.ru Gerasimov Evgeniy Yu., PhD, researcher at the Laboratory of Structural Research Methods of the Boreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences (Novosibirsk, Russian Federation). E-mail: gerasimov@catalysis.ru
Ayusheev Artemiy B., PhD, the leading expert of the Department of projects’ management at the Board of new products and technologies, «NIOST» Ltd. E-mail: a.ayusheev@gmail.com
Podyacheva Olga Yu., Dr, senior researcher in the Laboratory of Ecological Catalysis of the Boreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences (Novosibirsk, Russian Federation), senior researcher in the Laboratory of Synthesis and Physic-Chemical Investigations of New Composite Materials in the Novosibirsk National Research State University. E-mail: pod@catalysis.ru Taran Oxana P., Professor, leading scientist in the Laboratory of Catalytic Methods of Solar Energy Transformation of the Boreskov Institute of Catalysis, Siberian Branch of the Russian Academy of Sciences (Novosibirsk, Russian Federation), professor of Department of Engineering Issues of Ecology of Novosibirsk State Technical University (Novosibirsk, Russian Federation). E-mail: oxanap@catalysis.ru