

«клинические рекомендации «нарушения митохондриального бета-окисления жирных кислот»
(утв. минздравом россии)
МИНИСТЕРСТВО ЗДРАВООХРАНЕНИЯ РОССИЙСКОЙ ФЕДЕРАЦИИ
КЛИНИЧЕСКИЕ РЕКОМЕНДАЦИИ
НАРУШЕНИЯ МИТОХОНДРИАЛЬНОГО -ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
Кодирование по Международной статистической классификации болезней и проблем, связанных со здоровьем: E71.3
Год утверждения (частота пересмотра): 2021
Возрастная категория: Взрослые, Дети
Год окончания действия: 2023
ID: 694
Разработчик клинической рекомендации
— Ассоциация медицинских генетиков
— Союз педиатров России
— Общероссийская общественная организация содействия развитию неонатологии «Российское общество неонатологов»
Одобрено Научно-практическим Советом Минздрава РФ
Список сокращений
АЛТ — аланинаминотрансфераза;
АСТ — аспартатаминотрансфераза;
ацил-КоA — кофермент A, связанный с цепью жирных кислот;
БАД — биологически активная добавка;
ГА2 — глутаровая ацидурия тип 2;
ДЦТ — длинноцепочечные жирные кислоты (триглицериды);
ЖК — жирные кислоты;
КТ — компьютерная томография;
КФК — креатинфосфокиназа;
МРТ — магнитно-резонансная томография;
НБО — наследственные болезни обмена веществ;
ПНЖК — полиненасыщенные незаменимые жирные кислоты;
ССС — сердечно-сосудистая система;
СЦТ — среднецепочечные жирные кислоты (триглицериды);
ТМС — тандемная масс-спектрометрия;
УЗИ — ультразвуковое исследование;
ЭКГ — электрокардиография;
ЭНМГ — электронейромиография;
CACT (Carnitine-acylcarnitine translocase) — карнитин ацилкарнитин транслоказа;
CPT1 (Carnitine palmitoyltransferase I) — карнитин пальмитоилтрансфераза 1;
CPT2 (Carnitine palmitoyltransferase II) — карнитин пальмитоилтрансфераза 2;
DHA (Docosahexaenoic acid) — докозагексаеновая кислота;
ETF (Electron transfer flavoprotein) — электронпереносящий флавопротеин;
ETF QO-ETF (Electron transfer flavoprotein ubiquinone oxidoreductase) — электронпереносящий флавопротеин-убиквинон оксидоредуктаза;
FAOD (Fatty acid oxidation disorders) — нарушения митохондриального -окисление жирных кислот;
HELLP — синдром-акроним основных составляющих синдрома: гемолиза, повреждения печеночной паренхимы и тромбоцитопении;
LCHAD (Long-chain 3-hydroxyacyl-CoA dehydrogenase) — длинноцепочечная гидроксил-КоA дегидрогеназа жирных кислот;
LCFA (Long chain fatty acids) — длинноцепочечные жирные кислоты;
LCT (Long-chain triglycerides) — длинноцепочечные триглицериды (длинноцепочечные жиры);
MCFA (Medium-chain fatty acids) — среднецепочечные жирные кислоты;
MADD (Multiple acyl-CoA dehydrogenation deficiency) — множественный дефицит ацил-КоA дегидрогеназ;
MCAD (Medium-chain acyl-CoA dehydrogenase deficiency) — среднецепочечная ацил-КоA дегидрогеназа жирных кислот;
MCT (Medium chain triglycerides) — среднецепочечные триглицериды;
OCTN2 (Organic cation/carnitine transporter 2) — транспортер карнитина плазматической мембраны;
SCAD (Short-chain acyl-CoA dehydrogenase) — короткоцепочечной ацил-КоA дегидрогеназа жирных кислот;
TFP (Mitochondrial Trifunctional Protein) — митохондриальный трифункциональный белок;
TFPD (Mitochondrial trifunctional protein deficiency) — дефицит митохондриального трифункционального белка;
VLCAD (Very long-chain acyl-coenzyme A dehydrogenase) — очень длинноцепочечная ацил-КоA дегидрогеназа жирных кислот.
Термины и определения
Ацилкарнитины — эфиры жирной кислоты и карнитина;
Катаболизм — процесс распада сложных веществ;
Рабдомиолиз — разрушение клеток мышечной ткани;
Синдром Рейе — заболевание, проявляющееся острой невоспалительной (токсической) энцефалопатией в сочетании с жировой дистрофией внутренних органов, преимущественно печени (острый микровезикулярный стеатоз);
HELLP-синдром — тяжелое осложнение беременности, для которого характерна триада признаков: гемолиз, повреждение печеночной паренхимы с повышением активности печеночных трансаминаз и тромбоцитопения.
1. Краткая информация по заболеванию или состоянию (группы заболеваний или состояний)
1.1 Определение заболевания или состояния (группы заболеваний или состояний)
Митохондриальное -окисление жирных кислот является одним из важных биохимических процессов для выработки энергии в митохондриях.
Наследственные нарушения митохондриального -окисления жирных кислот (FAOD) — группа моногенных заболеваний, связанных с нарушением митохондриального -окисления и транспорта карнитина и жирных кислот в митохондриях.
Для всех FAOD характерно нарушение продукции энергии. Клинические симптомы, таким образом, развиваются или усиливаются на фоне катаболических состояний, например, при интеркуррентных инфекциях или голодании. Наиболее часто затрагиваются органы, для которых жирные кислоты служат основным источником энергии, такие как сердце и скелетные мышцы [1, 2]. Возраст пациента на момент дебюта, спектр клинических симптомов и степень тяжести заболевания могут различаться [1, 3, 4, 5, 6].
1.2 Этиология и патогенез заболевания или состояния (группы заболеваний или состояний)
Жирные кислоты (ЖК) являются важным источником энергии особенно в скелетных мышцах, сердце и печени, и их энергетическая роль возрастает в условиях голодания. ЖК мобилизуются из жировой ткани и транспортируются в кровоток, будучи первично связанными с альбумином. Большинство тканей способно расщеплять ЖК до углекислого газа и воды. На поздних стадиях голодания они становятся преимущественным источником продукции энергии через их окисление в скелетных мышцах и использование для продукции кетоновых тел в печени. ЖК являются главным источником энергии для сердца: в норме 60 — 70% энергии, необходимой для сердечной мышцы, обеспечивается окислением ЖК, и эти цифры могут быть даже выше в определенных ситуациях, например, при голодании и диабете.
Для осуществления окисления ЖК необходима определенная последовательность биохимических реакций: 1. Транспорт жирных кислот в митохондрию (проникновение ЖК через наружную и внутреннюю мембрану митохондрий); 2. Поэтапное укорочение углеродной цепочки жирных кислот до получения конечного продукта — ацетил-КоA (рис. 1).
Известно 11 различных форм наследственных FAOD. Данные о первичном молекулярно-генетическом дефекте при наиболее частых заболеваниях из этой группы приведены в приложении Г1. Сравнительно недавно были выявлены новые заболевания из этой группы, связанные с мутациями в генах ACAD9 и ECHS1 [74].
Рис. 1 Транспорт и окисление жирных кислот в митохондрии [105]
TFP — митохондриальный трифункциональный белок; ПМ — плазматическая мембрана; CI, CIII — комплексы дыхательной цепи митохондрий; MitoOM — внешняя митохондриальная мембрана; MitoIM — внутренняя митохондриальная мембрана; ETF — электронпереносящий флавопротеин; CPT1 — карнитин пальмитоилтрансфераза 1; CACT — карнитин ацилкарнитин транслоказа; CPT2 — карнитин пальмитоилтрансфераза 2; LCFA — длинноцепочечные жирные кислоты; MCFA — среднецепочечные жирные кислоты; OCTN2 — транспортер карнитина плазматической мембраны; ETF QO-ETF (электронпереносящий флавопротеин)-дегидрогеназа.
Молекулярные механизмы патогенеза FAOD и многие клинические проявления этих болезней сходны. Большинство FAOD характеризуются некетотической гипогликемией после периода голодания, поэтому гипогликемию эти пациенты переносят особенно тяжело. Истощение энергетических ресурсов при невозможности использования ЖК и токсическое действие последних обуславливают возникновение Рейе-подобного синдрома. Для многих форм FAOD характерно поражение мышечной ткани с миопатией и/или кардиомиопатией, что связано как с истощением энергетических ресурсов, так и с токсическим действием субстратов блокированных ферментных реакций на мышечную ткань.
1.3 Эпидемиология заболевания или состояния (группы заболеваний или состояний)
После включения FAOD в программы массового скрининга новорожденных были уточнены данные о частоте этой группы наследственных болезней. Считается, что их суммарная частота составляет 1:9000, хотя между популяциями могут быть значительные отличия [7]. Одно из самых частых заболеваний из этой группы — недостаточность среднецепочечной дегидрогеназы жирных кислот (MCAD) — 1:14600 — 1:20 000. Данные о частоте заболеваний из этой группы приведены в приложении Г1.
1.4 Особенности кодирования заболевания или состояния (группы заболеваний или состояний) по Международной статистической классификации болезней и проблем, связанных со здоровьем
Согласно МКБ-10, заболевание относится к классу IV, болезням эндокринной системы, расстройству питания и нарушению обмена веществ, E71.3 — нарушения обмена жирных кислот.
1.5 Классификация заболевания или состояния (группы заболеваний или состояний)
Известны 11 основных форм FAOD. Все они связаны с нарушением функции белков, которые участвуют в метаболизме жирных кислот и карнитина (приложение Г1). Краткая клиническая характеристика отдельных форм приведена в приложении Г2. В зависимости от метаболического блока различают формы, связанные непосредственно с нарушениями ферментов окисления жирных кислот и формы, связанные с дефектами метаболизма и транспорта карнитина и жирных кислот.
По ведущему симптомокомплексу, выделяют следующие формы FAOD:
— системную с поражением сердца и печени;
— печеночную;
— миопатическую.
По срокам появления первых признаков различают формы:
— неонатальную;
— детскую с манифестацией в первые два года жизни;
— позднюю.
1.6 Клиническая картина заболевания или состояния (группы заболеваний или состояний)
При всех формах FAOD выделяют разные по срокам манифестации варианты заболевания.
Общие клинические проявления нарушений окисления жирных кислот
FAOD имеют много общих клинических проявлений. Обычно выделяют неонатальную (раннюю), детскую (инфантильную) и позднюю (взрослую) формы болезни. Ранняя форма отличается особенной тяжестью, метаболические нарушения часто приводят к нарушению сознания, хотя коматозные состояния могут возникнуть и при детской, и при поздней формах.
Основные клинические проявления: неврологические нарушения в виде угнетения сознания вплоть до сомноленции и комы, общей вялости, повторная рвота, увеличение размеров печени, прогрессирующее поражение сердца в виде кардиомиопатии или нарушения сердечного ритма. Дети могут отставать в психомоторном развитии. У пациентов более старшего возраста доминируют миопатия и поражение миокарда, характерны приступы мышечной боли, рабдомиолиза и миоглобинурии, проявляющиеся изменением цвета мочи до коричневато-бурой окраски.
Характерной чертой FAOD является то, что клинические симптомы развиваются или усиливаются на фоне катаболических состояний, например, при продолжительной физической нагрузке, интеркуррентных инфекциях или голодании, когда происходит высвобождение эндогенных липидов. Наиболее часто затрагиваются органы, для которых длинноцепочечные жирные кислоты служат основным источником энергии, такие как сердце и скелетные мышцы [3, 8].
Частыми симптомами FAOD являются некетотическая гипогликемия, кардиомиопатия и миопатия, эти симптомы могут проявляться как изолированно, так и в сочетании.
Нарушения сердечного ритма и кардиомиопатия, наиболее часто проявляются в неонатальном периоде и раннем детстве, но могут развиться в период метаболического криза и в более позднем возрасте.
В раннем детстве поражение сердца может привести к летальному исходу. Довольно часто наблюдается гипертрофия левого желудочка, в тяжелых случаях — дилатационная кардиомиопатия со снижением фракции выброса. Иногда наблюдается кардиомиопатия в сочетании с выпотом в перикард [9, 10].
Нарушения сердечного ритма описаны практически при всех FAOD и встречаются в сочетании с кардиомиопатией или изолированно [11].
Миопатия очень частый симптом при нарушениях FAOD. Пациенты могут предъявлять жалобы на миалгию, быструю утомляемость, непереносимость физической нагрузки. Чаще всего миопатия манифестирует в подростковом возрасте, на 2 — 3 десятилетии жизни, но может быть ведущим симптомом и ранее. Иногда миопатия может провоцироваться интенсивными физическими упражнениями, но также может наблюдаться после анестезии и вирусной инфекции [1 — 5].
Гепатомегалия возникает вследствие накопления в печени жирных кислот с формированием жировой дистрофии печени [25]. У пациентов с недостаточностью LCHAD и TFP может наблюдаться клиническая картина периферической полиневропатии, причина развития которой до конца не известна [6, 113, 114].
У пациентов с дефицитом LCHAD (и с дефицитом TFP, но реже) развивается пигментная дегенерация сетчатки [115 — 117].
Наиболее частые клинические симптомы при различных формах FAOD приведены в приложении Г2а.
Особенности клинических проявлений отдельных форм FAOD приведены ниже.
Нарушения транспорта и метаболизма карнитина и жирных кислот
В группу входит несколько заболеваний, различающихся по срокам манифестации, тяжести течения и биохимическим характеристикам.
Транспорт длинноцепочечных ацил-КоA в митохондрии осуществляется с помощью карнитинового цикла, включающего 4 белка: 1) карнитиновый транспортер плазматической мембраны (OCTN2), обеспечивающий внутриклеточную доставку карнитина; 2) карнитин-пальмитоилтрансферазу 1 (CPT1) на внешней мембране митохондрий, которая переносит ацильные остатки ЖК от КоA к карнитину; 3) карнитин/ацилкарнитин транслоказу (CACT), обеспечивающую челночный транспорт ацилкарнитинов через внутреннюю мембрану в обмен на свободный карнитин; 4) карнитин-пальмитоилтрансферазу 2 (CPT2), которая переносит на внутренней мембране митохондрий ацильные остатки ЖК от карнитина к КоA. Мутации соответствующих генов, кодирующих данные белки, приводят к наследственным нарушениям транспорта карнитина и жирных кислот.
Системный дефицит карнитина
Аутосомно-рецессивное заболевание, связанное с мутациями гена SLC22A5, кодирующего транспортный белок — переносчик карнитина (OCTN2). Сроки манифестации заболевания от 1 месяца до 7 лет (в среднем 2 года).
У детей раннего возраста основное проявление системного дефицита карнитина — гипокетотическая гипогликемическая энцефалопатия (вялость, сонливость, повторная рвота), имеющая приступообразное течение. Отмечается увеличение размеров печени. Нередко у таких пациентов развивается синдром Рейе с гипераммониемией и повышением уровня трансаминаз. Также наблюдаются повышение уровней мочевой кислоты и КФК. Как правило, дети умеренно отстают в психомоторном развитии. Позже (как правило, в возрасте 2 — 3 лет) присоединяется прогрессирующая гипертрофическая или дилатационная кардиомиопатия в сочетании с нарушениями сердечного ритма, которые в отсутствие лечения неизбежно ведут к смерти от сердечной недостаточности и синдрому внезапной смерти. Дети часто болеют интеркуррентными заболеваниями [12, 13, 14]. Необычной манифестацией заболевания во младенческом возрасте является клиническая картина периферической полиневропатии с отсутствием сухожильных рефлексов и грубой задержкой психомоторного развития [118].
У пациентов более старшего возраста доминируют признаки скелетной миопатии и поражения миокарда. В мышечном биоптате у пациентов регистрируют накопление жиров. Нередко наблюдаются нарушения со стороны желудочно-кишечного тракта: боли в животе, диарея. В некоторых случаях наблюдалась периферическая невропатия верхних и нижних конечностей [17, 18]. При своевременной диагностике и назначении терапии наблюдается полное исчезновение клинической симптоматики.
Взрослые при данном заболевании могут быть асимптомными или отмечаются легкие симптомы в виде снижения выносливости, легкой утомляемости и плохой переносимости длительных голодных промежутков [146, 147]
Дефицит карнитин-ацилкарнитинтранслоказы
Аутосомно-рецессивное заболевание, связанное с мутациями гена SLC25A20, кодирующего карнитин-ацилкарнитинтранслоказу. Довольно редкое заболевание, в литературе описано не более 60 случаев [95, 104]. Начало заболевания — в неонатальный период (первые недели жизни) или у детей раннего возраста (на 1 году жизни). Заболевание манифестирует острой сердечно-легочной недостаточностью (коллапс) или желудочковыми аритмиями, мышечной слабостью и гипотонией. Наблюдается гипокетотическая гипогликемия, гипераммониемия. Синдром внезапной смерти младенца наблюдается в возрасте от 4-х дней до 24 месяцев. Наиболее частыми начальными симптомами являются: гепатомегалия (34%), аритмия и/или брадикардия (32%) и дыхательная недостаточность (30%). Гипокетотическая гипогликемия (68%) и гипераммониемия (54%) наиболее частые лабораторные изменения. Сердечная симптоматика включает гипертрофическую или дилатационную кардиомиопатию в сочетании с разнообразными нарушениями сердечного ритма. Печеночная недостаточность или умеренная гепатомегалия с метаболическим ацидозом и гипокетотической гипогликемией характерны для данного заболевания. Острая неврологическая симптоматика представлена нарушениями сознания (летаргией и комой) [23, 24, 102, 103]. Заболевание может манифестировать с мышечной слабости и задержки психомоторного развития. Также может наблюдаться повышение уровня КФК. При поздней манифестации отмечается меньшая тяжесть клинических проявлений [7, 27, 28].
Дефицит карнитинпальмитоилтрансферазы 1
Аутосомно-рецессивное заболевание, связанное с мутациями гена CPT1A, кодирующего карнитинпальмитоилтрансферазу 1, локализованную в печени.
Начало заболевания — обычно на первом году жизни, или в младенчестве. Обычно у пациентов отмечается незначительное увеличение печени, и заболевание протекает в виде острых метаболических приступов, как Рейе-подобный синдром, сопровождающихся тахипноэ, рвотой, отказом от еды, судорогами, летаргией, комой. Лабораторно выявляют повышение уровня трансаминаз, удлинением протромбинового и тромбопластинового времени. Нередко заболевание протекает под маской сепсиса
Триггерными факторами могут быть голодание, интеркуррентные инфекции, приводящие к гипокетотической/некетотической гипогликемии. На высоте приступа отмечается метаболический ацидоз. Приступ может привести к внезапной смерти.
Может наблюдаться задержка психомоторного развития. В единичных случаях сообщалось о скелетной миопатии, кардиомиопатии. Иногда развивается почечно-тубулярный ацидоз, который имеет преходящий характер и часто сочетается с миопатическим синдромом и повышением уровня КФК [148]. У женщин могут отмечаться тяжелые осложнения во время беременности — HELLP синдром (гемолиз, повышение печеночных трансаминаз, тромбоцитопения) [19].
Когнитивный дефицит у пациентов зависит от тяжести перенесенных гипогликемических состояний [119].
Дефицит карнитинпальмитоилтрансферазы 2
Аутосомно-рецессивное заболевание, связанное с мутациями гена CPT2. Выделяют три клинические формы патологии в зависимости от возраста манифестации: неонатальную, младенческую (инфантильную) и позднюю (мышечную) [13].
Течение заболевания острое или прогредиентное. Начало заболевания — неонатальный период. У пациентов с неонатальной формой наблюдается респираторный дистресс-синдром, кардиомиопатия, нарушение ритма и сердечной проводимости сердца [120, 121] гепатомегалия, печеночная недостаточность, энцефалопатия. Без лечения смерть от тяжелой сердечной аритмии и полиорганной недостаточности наступает в первые недели жизни. Встречаются дизморфии: скошенный лоб, микроцефалия, высокое небо, аномальные ушные раковины, длинные конусовидные пальцы, контрактуры, гипоплазия ногтей [31, 32]. Частым симптомом заболевания является увеличение размеров и поликистоз почек [122].
Младенческая манифестация данного заболевания потенциально относится к фатальным формам болезни. Младенческая форма заболевания чаще всего манифестирует в возрасте от 6 месяцев до двух лет в виде возникновения некетотической гипогликемии, увеличения печени, повышения уровня креатининфосфокиназы и аммония в крови особенно после длительного голодного промежутка. Так же у этих пациентов может развиваться метаболическая энцефалопатия с развитием эпилептических приступов, респираторного дистресс синдрома и метаболического ацидоза. Иногда наблюдается кардиомиопатия и аритмия.
Поздняя мышечная форма встречается чаще и характеризуется интермиттирующим течением и возрастом начала заболевания — на 2 — 3 десятилетии жизни. Заболевание манифестирует приступами мышечных болей, мышечной слабости, пароксизмальной миоглобинурией, как реакция на физическую нагрузку (непереносимость физических нагрузок). В редких случаях эти приступы наступают как реакция на голодание, интеркуррентные инфекции, эмоциональные стрессы, охлаждение. Приступы острой декомпенсации или дисфункция сердечной мышцы наблюдаются редко.
Почечная недостаточность вследствие миоглобинурии выявлена у 25% пациентов. Это заболевание является самой частой причиной наследственной миоглобинурии. Течение заболевания относительно доброкачественное [23, 24, 106].
Нарушения митохондриального -окисления жирных кислот
В митохондриальном матриксе ацил-КоA эфиры ЖК входят в -окисление, которое переносит электроны для синтеза АТФ (дыхательная цепь митохондрий). В результате -окисления ЖК последовательно расщепляются до ацетил-КоA, который затем превращается в кетоновые тела в печени. Первая реакция дегидрогенизации осуществляется ацил-КоA-дегидрогеназами. В окислении ЖК принимают участие короткоцепочечная, среднецепочечная, длинноцепочечная и очень длинноцепочечная ацил-КоA-дегидрогеназы (в английской транскрипции — SCAD, MCAD, LCAD и VLCAD, соответственно). Субстратная специфичность всех перечисленных ферментов различна и только частично перекрывается.
Дефицит ацил-КоA дегидрогеназы жирных кислот с короткой углеродной цепью.
Аутосомно-рецессивное состояние, связанное с мутациями гена ACADS, кодирующего ацил-КоA дегидрогеназу жирных кислот с короткой углеродной цепью (SCAD).
В многих случаях отмечают бессимптомное течение, поэтому SCAD теперь рассматривается как биохимический фенотип, а не заболевание. Большинство детей со SCAD были выявлены при проведении скрининга новорожденных, и подавляющее большинство из них остаются бессимптомными на протяжении нескольких лет.
Раньше выделяли тяжелую и легкую форму болезни [17, 53]. Впоследствии было обнаружено, что наличие в гомозиготном состоянии или в компаунд-гетерозиготном состоянии нескольких распространенных полиморфизмов приводит к снижению активности фермента SCAD и выявлению биохимических изменений в спектре ацилкарнитинов, но не имеет каких-либо физиологических последствий [75, 76].
Дефицит короткоцепочечной 3-гидроксиацил-КоA дегидрогеназы дефицит (SCHAD)
Аутосомно-рецессивное заболевание, связанное с мутациями гена HADH, кодирующем короткоцепочечную 3-гидроксиацил-КоA дегидрогеназу жирных кислот (SCHAD).
Заболевание в большинстве случаев манифестирует на первом году жизни, но возможен и более поздний возраст начала симптомов. Описано около десяти случаев дефицита SCHAD в литературе. Основные клинические проявления этого метаболического заболевания отличаются от других наследственных дефектов -окисления жирных кислот, поскольку при нем гипогликемия связана с гиперинсулинизмом. Также у пациентов отмечается миоглобинурия (в подростковом возрасте), гипокетотическая гипогликемическая энцефалопатия, гипертрофическая/дилатационная кардиомиопатия, мышечная гипотония, стеатоз печени, печеночная недостаточность. Описаны несколько случаев внезапной смерти. Стресс, голодание могут провоцировать симптомы.
Позднее установление диагноза может привести к формированию эпилепсии и умственной отсталости из-за повторяющейся гипогликемии [77].
SCHAD следует классифицировать как синдром врожденного гиперинсулинизма, поскольку у пациентов с доказанными мутациями HADH может наблюдаться рецидивирующая гипогликемия с гиперинсулинизмом, чувствительным к диазоксиду [77 — 79].
Другими частыми симптомами является задержка речевого развития, мышечная гипотония. Нередко отмечаются эпилептические приступы, миопатия, задержка физического развития, нарушения вскармливания. Реже встречаются дисморфичные черты, кардиомиопатия [75].
Дефицит ацил-CoA-дегидрогеназы среднецепочечных жирных кислот
Аутосомно-рецессивное заболевание, связанное с мутациями гена ACADM, кодирующего ацил-CoA-дегидрогеназу среднецепочечных жирных кислот (MCAD).
Заболевание в большинстве случаев манифестирует в возрасте от 3-х до 24-х месяцев. Нередко (от 10 до 40% случаев) заболевание начинается в периоде новорожденности. Приступы протекают тяжело и могут заканчиваться летально. В 5% случаев смерть детей происходит в первые дни жизни. Около 20% пациентов умирают до установления диагноза [11, 17]. В большинстве случаев основными проявлениями являются гипокетотическая гипогликемия, которая может сочетаться с лактат-ацидозом и гипераммониемией. Клинические проявления включают синдром угнетения ЦНС, сонливость и рвоту на фоне инфекционных заболеваний, голодания или при оперативных вмешательствах. У некоторых пациентов отмечается метаболический криз, несмотря на избыточное накопление кетоновых тел и нормальную концентрацию глюкозы в крови. Изредка пациенты демонстрируют во время приступа «парадоксально» выраженный кетоз. В период криза отмечается сонливость, рвота, дыхательная недостаточность, судороги, гепатомегалия, а также быстрое развитие сердечной недостаточности. Повреждение головного мозга, которое может возникнуть во время метаболического криза, увеличивает риск отдаленных неврологических нарушений. Может наблюдаться специфическая полиморфная желудочковая тахикардия в виде характерных изменений на ЭКГ в виде torsades de pointes (фр. — «скрученные шнурки»), или желудочковая тахикардия типа «пируэт». Направление комплексов QRS меняется циклически: несколько комплексов направлено вниз, затем в том же отведении — вверх. При объективном обследовании определяется увеличение печени, синдром цитолиза (Рейе-подобный синдром). Межприступный период протекает благоприятно, у 80% пациентов отмечается нормальное нервно-психическое развитие с полным отсутствием других признаков патологии [15, 17, 48, 49].
Дефицит ацил-КоA дегидрогеназы жирных кислот с очень длинной углеродной цепью
Аутосомно-рецессивное заболевание, связанное с мутациями гена ACADVL, кодирующего ацил-КоA дегидрогеназу жирных кислот с очень длинной углеродной цепью (VLCAD).
По срокам появления первых признаков выделяют три формы заболевания: неонатальную, детскую и позднюю. При неонатальной и детской формах различают следующие несколько клинических фенотипов [8, 13, 36, 37].
Наиболее распространенной формой заболевания является тяжелая младенческая форма (сердечная и полиорганная формы) с ранней манифестацией и высокой смертностью. При этих формах отмечается почти нулевая ферментативная активность, а клинически превалирует поражение сердца в виде (гипертрофической или дилатационной кардиомиопатии, нарушения сердечного ритма) и высокой летальностью на первом году жизни [123]; а также мышечной слабостью и гипотонией на фоне гипераммониемии, некетотической/гипокетотической гипогликемии и лактат-ацидозом. Острая неврологическая симптоматика представлена летаргией и комой. Синдром внезапной смерти младенца в неонатальный период или на первом году жизни характерен для данной формы [131].
Более легкая форма заболевания с такой же ранней манифестацией протекает как Рейе синдром. Клиника в виде острых приступов, сопровождающихся рвотой, вялым сосанием/отказом от пищи, мышечной гипотонией, гипорефлексией/арефлексией, тахипноэ, апноэ, летаргией, комой, развивается как реакция на голодание или вирусные инфекции, расстройства желудочно-кишечного тракта, приводящие к некетотической гипогликемии. На высоте приступа отмечается метаболический лактат-ацидоз, гиперурикемия, гепатомегалия, мышечная слабость. Приступ может привести к внезапной смерти. В ряде случаев гипогликемические приступы сопровождаются судорогами, диареей, миалгией, миоглобинурией, проксимальной миопатией. Энцефалопатия может наблюдаться даже в отсутствие генерализованных метаболических расстройств. После множественных приступов может развиться умственная отсталость. Несмотря на относительную легкость купирования приступов прогноз заболевания остается неопределенным, тогда как при неонатальной кардиомиопатической форме он неблагоприятен.
Поздняя форма (около 15% случаев) манифестирует обычно на втором-третьем десятилетии жизни непереносимостью физических нагрузок, миалгией, миоглобинурией и подъемом КФК в ответ на физическое переутомление и голод. Может развиться прогрессирующая проксимальная миопатия [38].
Дефицит митохондриального трифункционального белка (TFP)/недостаточность длинноцепочечной 3-OH ацил КоA дегидрогеназы жирных кислот (LCHAD)/недостаточность длинноцепочечной 3-кето-КоA тиолазы (LCKAT).
Митохондриальный трифункциональный белок (TFP) — мультиферментный комплекс, состоящий из 3 белков. Наиболее часто встречается недостаточность длинноцепочечной 3-OH ацил КоA дегидрогеназы жирных кислот (LCHAD), крайне редко недостаточность длинноцепочечной 3-кето-КоA тиолазы (LCKAT) [125].
Мутации в 2 генах приводят к данному заболеванию — HADHA, HADHB. Эти гены кодируют и субъединицы TFP. Мутации в этих генах могут приводить к генерализованной недостаточности TFP, вызывая его нестабильность, а также мутации в гене HADHA являются причиной изолированной недостаточности LCHAD, а мутации в HADHB гене — изолированной недостаточности LCKAT.
Клинический фенотип изолированной недостаточности LCHAD исключительно гетерогенен, напоминая по своим проявлениям недостаточность VLCAD. Также клинические фенотипы условно подразделяются на тяжелую неонатальную кардиомиопатическую форму (1), форму, преимущественно протекающую как Рейе синдром (2) в сочетании с поражением печени, легкую форму (3) с поздней манифестацией и преимущественным поражением скелетных мышц. Течение заболевания острое (1) и интермитирующее или прогредиентное (2 и 3).
Клинические проявления болезни в неонатальном периоде наиболее тяжелые, как правило приводящие к летальному исходу: кардиомиопатия, стеатоз гепатоцитов, мышечная гипотония, невропатия и сепсис-подобное состояние.
В раннем детском возрасте: характерна острая манифестация (рвота, общая мышечная гипотония, тонико-клонические судороги, вялость, сонливость, дыхательные расстройства, коматозные состояния). У многих детей наблюдается сочетание гипертрофической кардиомиопатии, гепатомегалии, мышечной гипотонии и слабости. При аускультации сердца выслушиваются глухие тоны, ритм галопа, при исследовании обращает внимание кардиомегалия, гипертрофия левого желудочка, нарушение систолической функции. Описана также перикардиальная эффузия. Характерна некетотическая/гипокетотическая гипогликемия как реакция на голодание.
Наиболее часто встречается форма заболевания с возрастом начала в первые 2 года жизни (2 дня — 2 года). У большинства пациентов заболевание манифестирует как Рейе синдром. Клиника в виде острых и, как правило, множественных приступов, сопровождающихся рвотой, вялым сосанием/отказом от пищи, мышечной гипотонией, гепатомегалией, гипорефлексией/арефлексией, тахипноэ, летаргией, комой, развивается как реакция на голодание или вирусные инфекции, расстройства желудочно-кишечного тракта, приводящие к некетотической гипогликемии. Приступ может привести к внезапной смерти. У некоторых пациентов отмечается прогредиентное течение заболевания в виде прогрессирующей кардиомиопатии, мышечной гипотонии и гепатомегалии. У всех пациентов выявляется лактат-ацидоз и высокая активность КФК.
В старшем возрасте у пациентов могут появиться приступы мышечных болей и миоглобинурии. У некоторых пациентов с поздней манифестацией развивается клиническая форма заболевания, сходная с мышечной формой CPT2 недостаточности. Эта форма манифестирует обычно на втором-третьем десятилетии жизни (но может манифестировать и раньше) непереносимостью физических нагрузок, миалгией, миоглобинурией и подъемом КФК в ответ на физическое переутомление и голод. Однако, для этих пациентов характерно сочетание этой клинической симптоматики с периферической невропатией и дисфункцией печени.
Поражение печени является одной из наиболее характерных черт LCHAD недостаточности и представлено гепатомегалией, синдромом цитолиза, в редких случаях — острой холестатической желтухой и печеночной недостаточностью с массивным тотальным некрозом печени [13, 41]. У женщин часто во время беременности может наблюдаться жировая дистрофия печени или HELLP-синдром [16, 80].
Клиническая картина дефекта TFP очень сходна с таковой при недостаточности LCHAD, но является более тяжелой и сопровождается ранним летальным исходом. Начало заболевания — в неонатальном периоде: прогрессирующая мышечная гипотония, летаргия, кормление затруднено, прогрессирующая сердечная недостаточность, синдром внезапной смерти младенца (в возрасте до 2-х месяцев). Очень тяжелая генерализованная неонатальная гипотония, клинически неотличимая от врожденной миопатии, была манифестирующим симптомом у 5 пациентов. В одном случае отмечалась поздняя манифестация заболевания (в 2,5 г) с относительно мягким интермиттирующим течением. Уникальный клинический фенотип описан у двух пациентов с манифестацией в виде прогрессирующей хронической полиневропатии и миопатии в отсутствие патологических изменений со стороны печени и сердечной мышцы.
Глутаровая ацидурия 2-го типа (множественный дефицит ацил-КоA дегидрогеназ)
Аутосомно-рецессивное заболевание, генетически гетерогенное, связанное с мутациями генов ETF-B, ETF-A, ETF-DH, которые кодируют альфа- и бета-субъединицы флавопротеина, являющегося переносчиком электронов (ETF), и ETF-кофермент Q-оксидоредуктазы. Нарушения функций любого из этих двух флавопротеинов приводит к нарушению окисления жирных кислот.
Выделяют несколько клинических форм ГА2: 1) тяжелую неонатальную форму с врожденными аномалиями; 2) тяжелую неонатальную форму без врожденных аномалий; 3) мягкую форму или этилмалоновую/адипиновую ацидурию. Кроме того, различают рибофлавин-резистентную и рибофлавин-чувствительную формы [13, 14].
При тяжелой неонатальной форме с врожденными аномалиями, как правило, ребенок рождается недоношенным. У детей с первых дней жизни развивается дыхательные нарушения (тахипноэ, апноэ), мышечная гипотония, вялость, сонливость вплоть до летаргии, рвота, увеличение печени и селезенки, необычный запах мочи («потных ног»), тяжелая анемия. Черты лица дизморфические, напоминающие таковые при синдроме Цельвегера: высокий лоб, гипертелоризм, гипоплазия лица по средней линии, диспластичные низко посаженные ушные раковины. Характерны врожденные пороки развития: мышечные дефекты передней брюшной стенки, гипоспадия, искривление полового члена, «стопа-качалка». Смерть в неонатальный период (в первую неделю жизни) на фоне летаргии и комы.
Неонатальная форма без врожденных аномалий, так же, как и предыдущая, отличается ранней манифестацией, тяжелым течением и нередко ведет к гибели. Дети отстают в психомоторном и физическом развитии. Характерно развитие Рейе-подобного синдрома и кардиомиопатии. Течение заболевания приступообразное. В некоторых случаях болезнь при отсутствии явных клинических проявлений может явиться причиной внезапной детской смерти в раннем возрасте [6].
Поздняя форма отличается более легким течением. Основными клиническими признаками являются: тошнота, рвота, увеличение печени, желтуха, мышечная слабость и гипотония. Умственное развитие не страдает. При поздней форме заболевания приступы гипокетотической гипогликемии протекают нетяжело, обычно не сопровождаются метаболическим ацидозом и гипераммониемией.
У части пациентов наблюдается улучшение состояния при назначении рибофлавина (биологическая активная добавка) [143]. Биохимические маркеры ГА2 могут совпадать с изменениями, выявляемыми при дефекте транспортера рибофлавина, которые характеризуются значительными клиническими улучшениями при назначении терапевтических доз рибофлавина (биологическая активная добавка). Краткие клинические описания этих заболеваний приведены ниже.
Вторичные нарушения митохондриального -окисления жирных кислот/сходные с ГА2 другие наследственные заболевания
Известны ряд наследственных заболеваний, при которых наблюдается изменение ацилкарнитинов, сходные с нарушениями -окисления жирных кислот.
Дефицит переносчика рибофлавина
Синдром Брауна-Виалетто-Ван Лаэра (BVVL) (MIM211530) — редкое неврологическое заболевание, которое проявляется нейросенсорной тугоухостью, бульбарным параличом и дыхательными нарушениями. Возраст начала варьирует от младенчества до взрослого [124]. Клинические признаки и симптомы BVVL сходны с болезнью Фацио-Лонде (MIM211500), которое первоначально рассматривалась как отдельная форма, но теперь известно, что это аллельный вариант BVVL [124]. К данным заболеваниям приводят мутации в гене SLC52A3, который кодирует переносчик рибофлавина (RFVT3), или в гене SLC52A2, кодирующем другой переносчик рибофлавина плазматической мембраны (RFVT2). Клинические проявления у пациентов с дефицитом RFVT2 и RFVT3 практически неотличимы. У многих пациентов с этой патологией выявляют измененный профиль ацилкарнитинов в крови, сходный с ГА2 [125].
Для терапии синдрома Брауна-Виалетто-Ван Лаэра и болезни Фацио-Лонда высокоэффективен рибофлавин (биологическая активная добавка) в высоких дозах, что требует проведения исследований для дифференциальной диагностики данных нарушений с ГА2 [125].
Дефицит FAD-синтазы (FADS)
Дефицит FAD-синтазы (FADS) был впервые описан в 2022 году в 7 неродственных семьях, с неврологическими проявлениями и биохимическим профилем, указывающим на ГА2 [126]. При лечения этой патологии также применяют рибофлавин (биологическая активная добавка).
Дефицит митохондриального переносчика FAD
В 2022 году был описан дефицит митохондриального переносчика FAD у 14-летней девочки с непереносимостью физических нагрузок [127]. Лабораторные исследования выявили профиль, похожий на ГА2. Были выявлены мутации в гене SLC25A32, который кодирует белок из семейства транспротеров (митохондриального переносчика FAD). В 2022 году был описан второй пациент с данным нарушением, но более тяжелой клинической картиной (атаксия, дизартрия, миоклонус, мышечная слабость и непереносимостью физических нагрузок). Состояние пациента улучшилось после приема рибофлавина (биологическая активная добавка) [128].
Дифференциальная диагностика FAOD проводится с широком кругом заболеваний наследственной и экзогенной природы: митохондриальными болезнями, связанными с дефектами дыхательной цепи митохондрий, наследственными формами гипертрофических кардиомиопатий, нарушениями обмена углеводов, миопатиями, органическими ацидуриями и нарушениями цикла мочевины.
Следует проводить дифференциальную диагностику с гипоксическими поражениями нервной системы, внутриутробными инфекциями, а также с кардиомиопатиями, гепатитами различного происхождения [4, 9, 10, 11].
2. Диагностика заболевания или состояния (группы заболеваний или состояний) медицинские показания и противопоказания к применению методов диагностики
Диагноз устанавливается на основании совокупности клинических данных, результатов лабораторного исследования [5, 7].
2.1 Жалобы и анамнез
При сборе анамнеза и жалоб следует обратить внимание на следующие жалобы и анамнестические события:
— Отягощенный семейный анамнез (сходные симптомы у родных братьев и сестер пробанда, близкородственный брак);
— Случаи внезапной детской смерти;
— HELLP — синдром или жировая дистрофия печени у женщины во время беременности;
— Рвота (в любом возрасте);
— Судороги (в любом возрасте);
— Изменение цвета мочи (красновато-бурая моча) (в любом возрасте);
— Эпизоды нарушения сознания (в любом возрасте);
— Эпизоды нарушений сердечного ритма (в любом возрасте);
— Непереносимость длительных голодных промежутков;
— Непереносимость физической нагрузки (взрослые и дети старшего возраста);
— Мышечные боли (взрослые и дети старшего возраста);
— Быстрая утомляемость (взрослые и дети старшего возраста);
— Эпизоды гипокетотической гипогликемии в анамнезе.
Жалобы и анамнез также описаны в разделе «клиническая картина».
2.2 Физикальное обследование
При осмотре необходимо обратить внимание на основные клинические проявления FAOD:
— Сонливость, вялость;
— Эпизоды нарушения сознания;
— Судороги;
— Мышечная гипотония;
— Мышечная слабость;
— Боли в мышцах при пальпации;
— Увеличение размеров печени;
— Нарушение сердечного ритма;
— Признаки сердечной недостаточности.
Подробно данные физикального обследования описаны в разделе «клиническая картина».
2.3 Лабораторные диагностические исследования
Основные лабораторные методы подтверждения диагноза FAOD включают определение ацилкарнитинов методом ТМС, анализ органических кислот мочи и молекулярно-генетические исследования генов, ответственных за -окисление жирных кислот, метаболизм и транспорт карнитина. Данные исследования проводятся в специализированных генетических лабораториях.
Рекомендовано Исследование уровня свободного карнитина и ацилкарнитинов в крови всем пациентам с клиническими симптомами, характерными для FAOD [1, 105, 144].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Для каждой из форм FAOD характерен свой спектр ацилкарнитинов, приложение Г3. Низкий уровень свободного карнитина также может наблюдаться при ряде заболеваний из группы FAOD. Следует отметить, что, если уровень свободного карнитина низкий, повышение специфических ацилкарнитинов будет не выраженным и это может привести к ложноотрицательным результатам анализа. При выявлении профиля ацилкарнитинов, характерного для ГА2 необходимо проводить дифференциальную диагностику с дефектами метаболизма рибофлавина (см. Раздел Клиническая картина заболевания или состояния).
Правила забора крови на фильтр приведена в приложении Г4.
— Рекомендовано всем пациентам с клиническими симптомами характерными для FAOD определение содержания органических кислот в моче методом газовой хроматографии масс-спектрометрии для исключения других заболеваний из класса наследственных болезней обмена веществ и диагностики ГА2 [6, 13].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: В моче у пациентов с FAOD могут повышаться концентрации дикарбоновых кислот, лактата, глутаровой кислоты по сравнению с референсными значениями. В большинстве случаев эти изменения неспецифичны, за исключением ГА2 (приложение Г4).
— Рекомендовано всем пациентам при выявлении биохимических изменений, свидетельствующих в пользу FAOD исследование соответствующих генов для подтверждения диагноза и проведения медико-генетического консультирования семьи [9].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 4).
Комментарий: Новые варианты неизвестного значения, требуют функциональных исследований, например, на культуре кожных фибробластов, чтобы установить, являются ли они причиной заболевания.
— Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD общий (клинический) анализ крови развернутый (гемоглобин, количество эритроцитов, цветовой показатель, количество лейкоцитов, тромбоцитов, лейкоцитарная формула и скорость оседания эритроцитов) для оценки основных параметров кроветворения и наличия воспалительных процессов [1, 2, 5].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD проведение анализа крови биохимического общетерапевтического (содержание глюкозы, общего белка, белковых фракций, альбумин, C-реактивного белка, исследование уровня общего билирубина в крови, Исследование уровня свободного и связанного билирубина в крови, холестерина, триглицеридов, липопротеидов низкой и высокой плотности (исследование уровня липопротеинов в крови), щелочной фосфатазы, креатинина, мочевины, мочевой кислоты, аммония, аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), гаммаглютамилтрансферазы (ГГТ), креатинфосфокиназы (КФК), Исследование уровня/активности изоферментов креатинкиназы в крови, лактатдегидрогеназа (ЛДГ), кальция общего и ионизированного, натрия, калия, неорганического фосфора, железа, ферритина, магния, хлора, молочной кислоты), инсулина, показателей кислотно-основного равновесия (Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (pH) крови), исследование уровня молочной кислоты в крови (лактата) с целью оценки состояния печени, почек, баланса важнейших нутриентов при диетотерапии, кальциево-фосфорного обмена а также для оценки риска метаболической декомпенсации [12, 132].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: у пациентов с FAOD и наиболее значимо в период метаболической декомпенсации повышается уровень трансаминаз, креатинфосфокиназы, наблюдается гипогликемия и метаболический ацидоз. Перечень основных лабораторных исследований, которые могут помочь в оказании медицинской помощи при состоянии острой метаболической декомпенсации, приведены в приложении Г18.
— Рекомендуется определение лабораторных показателей (кислотно-щелочного состояния крови (Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (pH) крови), исследование уровня молочной кислоты в крови (лактата), глюкоза, электролиты (калий, натрий, кальций ионизированный, хлор) в период и при подозрении на метаболический криз пациентам с клиническими и/или биохимическими признаками FAOD с целью оценки состояния и своевременной коррекции терапии [12, 132].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 4).
— Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD общий (клинический) анализ мочи, определение кетоновых тел для оценки состояния мочевыводящих путей и почек [1, 2, 5].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: в связи с высоким риском развития интеркуррентных инфекций у пациентов с FAOD рекомендуется проведение данного исследования не реже 5 раз в год
— Рекомендуется всем пациентам с патологией сердечно-сосудистой системы исследования уровня N-терминального фрагмента натрийуретического пропептида мозгового (NT-proBNP) в крови для своевременной диагностики сердечной недостаточности, дифференциальной диагностики с одышкой, вызванной респираторными нарушениями, для решения вопросов о старте/коррекции кардиотропной терапии [11, 133].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 4).
2.4 Инструментальные диагностические исследования
— Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD проведение ЭКГ и ЭхоКГ для оценки состояния сердца, при наличии нарушения ритма и проводимости проведение холтеровского мониторирования ЭКГ [11, 97, 109, 129].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 4).
Комментарии: регулярное проведение ЭКГ, Эхо-КГ, необходимо пациентам данной патологией, так как с раннего возраста у них отмечаются сердечно-сосудистые нарушения.
В соответствии с Приказом Минздрава России от 13 октября 2022 г. N 804н «Об утверждении номенклатуры медицинских услуг»: мониторирование электрокардиографических данных, Холтеровское мониторирование сердечного ритма, расшифровка, описание и интерпретация электрокардиографических данных,
Регистрация электрокардиограммы.
— Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD ультразвукового исследования (УЗИ) органов брюшной полости и почек для исключения/подтверждения патологии печени и почек [9, 12, 13, 15, 145].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Рекомендовано всем пациентам с клиническими и/или биохимическими признаками FAOD проведение исследования состояния глазного дна с целью выявления пигментной дегенерации сетчатки [1, 2, 5].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
2.5 Иные диагностические исследования
Консультации специалистов могут оказываться пациентам на разных этапах оказания медицинской помощи, в том числе в период диагностики заболевания.
— Рекомендуются при постановке диагноза пациентам с клиническими и/или биохимическими признаками FAOD применять мультидисциплинарный подход ввиду того, что заболевания характеризуются поражением многих органов и систем, требует комплексной терапии, что диктует необходимость совместного ведения пациента специалистами разных профилей [12, 13, 15, 145].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5)
Комментарии: показаны первичные консультации и повторные консультации врача-генетика, врача-офтальмолога, врача-невролога, врача-диетолога, врача-гастроэнтеролога, врача-эндокринолога, врача-кардиолога, врача-детского кардиолога, врача-нефролога, врача-педиатра/врача-терапевта/врача общей практики (семейного врача), а также врачей других специальностей пациентам с FAOD, имеющим нарушения функций соответствующих органов и систем.
3. Лечение, включая медикаментозную и немедикаментозную терапии, диетотерапию, обезболивание, медицинские показания и противопоказания к применению методов лечения
3.1 Общие рекомендации по диетотерапии пациентов с FAOD
Стратегия лечения пациентов с FAOD носит комплексный характер, в его основе лежит коррекция метаболических нарушений посредством диеты, которая заключается в снижении потребления пищевых жиров в качестве резервной составляющей тканевой биоэнергетики, минимизации катаболизма жирных кислот и уменьшении их значимости для восполнения энергозатрат клетки с обеспечением нормальных процессов анаболизма, роста и нутритивного статуса. Главная задача диетотерапии — это профилактика голодания, предупреждение гипогликемии и минимально допустимое снижение поступления с пищей патогенетически значимых жирных кислот и их источников.
Поступление достаточной энергии за счет углеводов является ключевым моментом в диетотерапии FAOD. Процесс анаболизма подавляет окисление жирных кислот и снижает уровень циркулирующих ацилкарнитинов. Расчет калорийности питания должен обеспечивать адекватное поступление энергии, препятствуя катаболизму, и в то же время не приводить к перекармливанию и к патологической прибавке веса. По результатам проведенных исследований у пациентов с FAOD были отмечены следующие особенности метаболизма [81]:
— более низкие энергозатраты;
— преимущественное окисление углеводов для получения энергии;
— низкая мышечная масса тела и большая жировая масса тела.
— Рекомендуется избегать длительного голодания всем пациентам с FAOD для предотвращения развития метаболических кризов [11 — 14, 145].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарий: в приложении Г5 суммированы рекомендации по продолжительности периодов голодания в соответствии с возрастом. Однако указанные значения применимы к здоровым детям и стабильным состояниям, когда в условиях нормокалорийного питания дневные запасы гликогена используются в период продолжительного ночного голодания для продукции глюкозы. Необходимо учитывать индивидуальные особенности ребенка, для некоторых пациентов требуются меньшие интервалы между приемами пищи для сохранения стабильного состояния.
Начиная с 8 — 12 месяцев, с целью предотвращения катаболизма в ночной период, можно давать перед сном кукурузный крахмал (1 г/кг/день (разведение крахмала в детской смеси или в воде 1:2), при регистрации гипогликемии в ночные часы возможно дополнительное введение крахмала в той же дозе) для обеспечения поступления адекватной энергии в течение ночи, данная тактика обычно используется только у пациентов с тяжелым течением заболевания, склонных к гипогликемии. В приложении Г6 приведены среднесуточные нормы физиологических потребностей в энергии для детей.
— Рекомендовано назначение диетотерапии всем пациентам с FAOD (кроме MCADD, SCADD, системной недостаточности карнитина) после установления точного диагноза с целью снижения рисков возникновения метаболической декомпенсации [11 — 14].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 4).
Комментарии: в зависимости от формы заболевания рекомендуются разные схемы лечения. В приложении Г7 приведены основные рекомендации по соотношению жиров и углеводов в диете.
3.1.1 Лечение пациентов с FAOD в период интеркуррентных инфекций
У пациентов с FAOD эпизоды метаболической декомпенсации могут наблюдаться при интеркуррентных заболеваниях, гастроэнтерите.
— Рекомендуется поддерживать высокое потребление углеводов во время любого метаболического стресса всем пациентам с FAOD для предотвращения развития метаболического криза [110].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5)
Комментарии: напитки с 20 — 25% раствором декстрозы или кукурузного крахмала (кукурузный крахмал — пациентам старше 8 месяцев жизни в разовой дозе 1 г/кг) следует начинать при первых признаках заболевания, а затем равномерно распределять в течение суток. Для тех пациентов, у которых установлен зонд проводится непрерывное энтеральное питание или повторный болюс питательного раствора, уже используемого ночью, может быть предложен в течение всего дня. В случаях клинического ухудшения с отказом от еды, рвотой, необходима госпитализация для проведения инфузионной терапии (Приложение Г9).
3.1.2 Рекомендации по лечению пациентов с FAOD в период метаболической декомпенсации
У всех пациентов с FAOD, независимо от наличия или отсутствия клинических симптомов, катаболические состояния требуют применения режима неотложной ситуации.
Состояние метаболического криза является показанием для госпитализации и проведения интенсивной, в том числе инфузионной терапии, которая должна начинаться незамедлительно. Тактика лечения детей в период криза включает дополнительное введение декстрозы** для энергетической поддержки и уменьшения интенсивности процессов катаболизма, коррекцию метаболического ацидоза, гипераммониемии и водно-электролитных нарушений, коррекцию диетотерапии.
Устранение гипогликемии и энергетической недостаточности имеет первостепенное значение для сохранения жизни и здоровья пациентов с FAOD.
— Рекомендуется внутривенное введение раствора декстрозы** под контролем ее уровня в крови пациентам с FAOD при развитии метаболического криза [5, 12, 52, 130, 150].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 4).
Комментарии: Внутривенное введение раствора декстрозы** из расчета: новорожденные и дети до 3-х лет 10 — 12 мг/кг/мин, от 3 до 10 лет 8 — 10 мг/кг/мин, старше 10 лет 5 — 8 мг/кг/мин. При снижение глюкозы ниже 2,5 ммоль/л в крови необходимо внутривенное струйное введение 10% — 25% декстрозы** из расчета 2 — 3 мл/кг, далее переходят на внутривенное капельное введение 10% декстрозы** со скоростью 7 — 9 мг/кг/мин до нормализации уровня глюкозы в крови.) Назначение декстрозы** не только восполняет тканевой энергетический дефицит, но и подавляет липолиз и снижает продукцию токсичных дериватов жирных кислот.
Неотложную внутривенную терапию следует начинать как можно раньше при более тяжелых состояниях, с высокой температурой, повторяющейся рвотой или тяжелым гастроэнтеритом.
Растворы для внутривенных инфузий, содержащие 10% декстрозу** (или более) следует использовать даже при нормальном уровне глюкозы в крови. Пациенты с FAOD полностью зависят от глюкозы для своих энергетических потребностей и ее запас может быстро истощаться. Если у пациента развивается значительная гипергликемия (например, уровень глюкозы в крови больше 250 мг/дл) с глюкозурией и имеются признаки метаболической декомпенсации, то следует рассмотреть возможность начала инфузии #инсулина (инсулин растворимый (человеческий генно-инженерный) **) вместо снижения скорости инфузии декстрозы**, способствуя анаболическому состоянию. Начальная доза для инфузии инсулина составляет 0,01 МЕ/кг/мин. Уровень инсулина следует титровать, чтобы поддерживать уровень глюкозы в крови между 100 и 150 мг/дл [153].
Следует отметить, что инфузия более концентрированного раствора декстрозы** (15 — 25%) при более низкой скорости инфузии посредством центрального катетера может потребоваться при недостаточной сердечной функции. Кроме того, следует соблюдать осторожность при прекращении внутривенной инфузии — необходимо медленно снижать скорость, чтобы избежать состояния реактивной гипогликемии.
— Рекомендуется коррекция метаболического ацидоза (при уровне бикарбонатов сыворотки крови < 16 мЭкв/л) пациентам с FAOD при развитии метаболического криза [5, 12, 52].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 4).
Комментарии: согласно клиническим рекомендациям по интенсивной терапии дефицит бикарбонатов купируется путем внутривенного введения щелочных растворов: натрия гидрокарбонат**. Натрия гидрокарбонат** применяется в виде 8,4% и 4,2% раствора для удобства перерасчета на ммоль NaHCO. Его дозировка (ммоль) определяется по формуле: (-BE) x масса тела (кг) x 0,3. Также пациентам может быть назначено щелочное питье — раствор соды из расчета 1/2 — 1 чайная ложка на 200 мл воды, щелочные минеральные воды.
В зависимости от тяжести состояния каждые 6 — 12 часов контролировать показатели кислотно-основного состояния крови, уровня натрия и калия в крови пациентам с FAOD при развитии метаболического криза [5, 12, 13, 14].
— Рекомендуется коррекция водно-электролитных нарушений пациентам с FAOD при развитии метаболического криза [5, 12, 145].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: при сохраняющейся гипогидратации проводится путем внутривенного введения натрия хлорида** 0,9%). Однако необходимо иметь в виду, что главным мероприятием в комплексе интенсивной терапии является введение растворов декстрозы и щелочных растворов.
Рекомендовано: назначение натрия бензоата (биологически активная добавка) при уровне аммиака в крови выше 200 мкмоль/л пациентам с FAOD при развитии метаболического криза [152, 154].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Натрия бензоат (биологически активная добавка) назначается из расчета от 250 мгкгв сутки. (максимально до 500 мг/кг/сут, если масса тела превышает 20 кг до 5,5 г/м2/сут)
Контроль уровня аммиака должен проводиться каждые 6 — 12 часов. Осуществляют обязательный контроль уровня натрия и калия в крови не реже 1 раза в 2 часа.
Согласно международным рекомендациям по лечению гипераммониемии показано внутривенное введение препаратов, связывающих аммиак, однако в настоящее время на территории Российской Федерации данные лекарственные средства не зарегистрированы. Поэтому все препараты, связывающие аммиак, являются препаратами off-label или не зарегистрированы и требуют оформления врачебной комиссии и согласия родителей/законных представителей и пациента с 15 лет. Прием данных препаратов при гипераммониемии необходим по жизненным показаниям
— Рекомендуется коррекция диетических мероприятий всем пациентам с FAOD в период метаболического криза с целью компенсации данного состояния [1, 105].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: При VLCAD, LCHAD диетические ограничения при кризе: полностью исключить потребление жиров на период острого криза (на 24 — 48 часов), далее вводить минимальное количество среднецепочечных жиров при сохранении высокой калорийности (не менее 100 — 115 ккал/кг) рациона в основном за счет углеводов; избегать голодания, когда прекращаются инфузии.
— Рекомендуется использовать приемлемый энтеральный способ кормления для пациента с FAOD для предотвращения развития катаболических состояний [1, 105].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: самостоятельно через рот, через зонд или гастростому.
Кормление через назогастральный зонд в рамках долгосрочной терапии, в целом, нежелательно, но рекомендуется в условиях заболевания на начальном этапе метаболического нарушения. В повседневной практике назначение декстрозы** или кукурузного крахмала через рот не требуется, а применяется только при угрозе возникновения или во время метаболического криза.
— В случае необходимости введения эпинефрина** всем пациентам с FAOD рекомендовано его введение вместе с 10% раствором декстрозы** [97, 151].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Эпинефрин** может стимулировать липолиз, поэтому при назначении этим детям следует добавлять декстрозу**
Международные клинические рекомендации по лечению разработаны только для отдельных форм FAOD — дефектах среднецепочечной дегидрогеназы жирных кислот и длинноцепочечной дегидрогеназы жирных кислот. Для других форм FAOD рекомендации по диетотерапии могут различаться в разных странах. Ниже приводятся основные рекомендации, которые по мнению рабочей группы являются наиболее полными.
3.2 Рекомендации по терапии отдельных форм FAOD
В зависимости от первичного метаболического дефекта подходы к терапии могут существенно различаться или иметь минорные различия.
3.2.1 Рекомендации по лечению системной недостаточности карнитина
— Не рекомендовано назначение специальной диетотерапии пациентам с системной недостаточностью карнитина, поскольку ведущим патогенетическим механизмом развития заболевания является дефицит карнитина [82].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: восполнение карнитина является основным методом лечения данной патологии. Также пациентам следует избегать перерывов в приеме пищи (см. общие рекомендации для FAOD и Приложение Г4).
— Рекомендовано назначение #левокарнитина в дозе 50 — 400 мг/кг/сутки, всем пациентам с подтвержденной первичной недостаточностью карнитина с целью снижение риска возникновения гипогликемических эпизодов [54, 82, 155].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: #Левокарнитин назначают в дозе 50 — 400 мг/кг/сутки, разделенной на три приема. Точная доза #левокарнитина должна быть скорректирована в зависимости от уровня карнитина в плазме. Концентрацию карнитина следует измерять как минимум через 4 ч после последнего приема препарата. Концентрация свободного карнитина у пациентов с первичным дефицитом карнитина сохраняется ниже нормы даже при его применении [14]. Не существует установленного «терапевтического» уровня, которого следует достигнуть и адекватной считается концентрация свободного карнитина > 20 мкМ. Уровень карнитина в плазме быстро падает после прекращения терапии #Левокарнитином и затем происходит истощение его запаса в тканях. Это объясняет, почему пациенты с первичным дефицитом карнитина, которые пропускают прием препарата, остаются бессимптомными несколько месяцев, что может привести к тому, что они прекращают прием #левокарнитина. Эти пациенты подвергаются риску внезапной смерти из-за остановки сердечной деятельности. Очень важно продолжать терапию ежедневно, чтобы избежать тяжелых нарушений со стороны сердца. Длительная терапия #Левокарнитином полностью купирует клинические проявления заболевания с нормализацией размеров сердца и ЭКГ в течение месяца с восстановлением мышечного тонуса, хотя тканевые (в мышечной ткани) концентрации карнитина остаются очень низкими (< 10% от N) в течение длительного времени.
Прием #Левокарнитином в высоких дозах может вызывать усиление моторики желудочно-кишечного тракта, диарею, и выработку триметиламина, что является причиной «рыбного запаха» от мочи и тела. Уменьшение дозы карнитина может уменьшить эти побочные эффекты. Если неприятный запах сохраняется, возможно назначение курса перорального приема #метронидазола** в дозе 10 мг/кг/сутки в течение 7 — 10 дней [82, 83, 155, 156].
3.2.2 Рекомендации по лечению дефицита карнитин-ацилкарнитин транслоказы
Клинические рекомендации, основанные на достаточном уровне доказательности и убедительности, для лечения и мониторинга этого конкретного заболевания в мире не разработаны. Были опубликованы только рекомендации, основанные на опыте экспертов по клиническому и биохимическому мониторингу пациентов с FAOD. Нет единого мнения относительно использования левокарнитина при этой форме болезни. Некоторые авторы не рекомендуют этого, поскольку накопление длинноцепочечных ацилкарнитинов может быть токсичным для сердечного ритма [84]. Тем не менее, другие авторы считают целесообразным применять его при эпизодах метаболической декомпенсации, для восстановления митохондриального пула коэнзима A [66, 85 — 89].
— Рекомендуется пациентам с установленным диагнозом CACT соблюдение низкожировой диеты (потребление жиров должно составлять 30% или менее от общей калорийности, 20% из них — MCT с низким содержанием C10 и 10% LCT (длинноцепочечные триглицериды) для предотвращения развития метаболического криза [66, 87 — 89].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: рекомендовано MCT с низким содержанием C10 в связи с тем, что жиры C10 требуют для окисления активной работы CACT [90].
— Рекомендуется пациентам с установленным диагнозом CACT избегать длительных перерывов в приеме пищи для предотвращения гипогликемических состояний [66, 87 — 89].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: не существует единого мнения о безопасных интервалах между кормлениями, в данных клинических рекомендациях нами были выбраны те, которые опубликованы в работе группы экспертов из европейских стран. Максимально допустимый безопасный интервал для разных возрастных групп приведен в Приложение Г6[66].
— Рекомендуются пациентам с установленным диагнозом CACT добавки незаменимых жирных кислот, с целью предотвращения их дефицита — линолевой (3 — 4%) и линоленовой (0,5 — 1%) — в соотношении от 5:1 до 10:1, их источником могут являются масло грецкого ореха, льняное, соевое [66, 87 — 89].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5)
Комментарии: Содержание кислот в разных маслах приведено в Приложение Г13[66].
— Рекомендуется пациентам с установленным диагнозом CACT в период интекуррентных инфекций и других состояний, при которых активируются механизмы катаболизма, применение перорально растворов декстрозы** или полимеров декстрозы в виде пищевой добавки (мальтодекстрин (декстринмальтоза)) и повышенное потребление углеводов [89].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: детальные рекомендации см. в разделе 3.1.2 Рекомендации по лечению пациентов с FAOD. В период метаболической декомпенсации снижение уровня длинноцепочечных ацилкарнитинов считается важным маркером эффективности лечения.
3.2.3 Рекомендации по рациону питания при дефиците карнитин пальмитоил-CoA трансферазы I, II
Клинические рекомендации, основанные на достаточном уровне доказательности и убедительности, для лечения и мониторинга дефицита CPT2 и CPT1 в мире не разработаны. Были опубликованы только рекомендации, основанные на опыте экспертов по клиническому и биохимическому мониторингу пациентов с FAOD.
— Рекомендуется пациентам с установленным диагнозом CPT2 и CPT1 избегать длительных перерывов в приеме пищи для предотвращения гипогликемических состояний [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: частое кормление каждые 3 — 4 часа рекомендуется, особенно для детей, учитывая их ограниченные запасы гликогена. Добавление кукурузного крахмала на ночь и в ночные кормления определяется уровнем гликемии и обеспечивает постоянный источник углеводов с медленным высвобождением для предотвращения гипогликемии во время сна. Дети более старшего возраста не должны иметь ночные перерывы в приеме пищи более 12 часов, а в период инфекционного заболевания перерывы в приеме пищи должны быть еще меньше. Взрослые пациенты должны быть проинформированы о рисках, связанных с голоданием, и они, и их лечащий врач должны знать о рисках во время хирургических операций и анестезии. Следует рекомендовать внутривенное введение декстрозы**, если необходимо голодать более 12 часов из-за болезни или медицинских процедур. Максимально допустимый безопасный интервал для разных возрастных групп приведен в Приложение Г5
— Рекомендуется диета с высоким содержанием углеводов (70% калорий) с низким содержанием жира (< 20% калорий) пациентам с установленным диагнозом дефицит CPT2 и CPT1 для предотвращения гипогликемических состояний и метаболической декомпенсации [91].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Рекомендуется в период интеркуррентных инфекций и других состояний, при которых активируются механизмы катаболизма, пациентам с установленным диагнозом дефицит CPT2 и CPT1 с целью предотвращения метаболической декомпенсации употреблять примерно треть от общего количества калорий в виде MCT [91].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: жирные кислоты C6 — C10 не требуют карнитинового челнока для входа в митохондрию, поэтому MCT может применяться в качестве основного источника энергии.
— С целью профилактики эпизодов миоглобинурии пациентам с установленным диагнозом CPT1, CPT2 рекомендуется избегать физических нагрузок во время интеркуррентных инфекций и при длительных перерывах в приеме пищи [91, 92].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Взрослым пациентам с установленным диагнозом CPT1, CPT2 в период метаболического криза, сопровождаемого рабдомиолизом и миоглобинурией рекомендуется применение стандартного протокола лечения этого осложнения, чтобы предотвратить острую почечную недостаточность [13, 92, 93].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: при приступе миоглобинурии проводят внутривенное введение 0,9% раствора натрия хлорида** и натрия гидрокарбоната ** (натрия хлорида** 110 ммоль/л, натрия гидрокарбоната** 40 ммоль/л) в 5% растворе декстрозы**, к которому добавляют 10 г #маннитола** на литр (из 10% или 15% раствора). Раствор следует вводить молодому взрослому человеку весом 75 кг из расчета 12 л/сут, чтобы получить диурез 8 л/сут и поддерживать pH выше 6,5. Этот терапевтический режим позволяет контролировать гиперкалиемию и ацидоз и, следовательно, может предотвратить острую почечную недостаточность. Если пациенты не реагируют на регидратацию, может потребоваться гемодиализ. Гемодиализ корректирует метаболический ацидоз и нарушения электролитов и способствует удалению мионекротических токсинов из плазмы. Диализ показан при почечной недостаточности, если у пациента анурия и диурез не восстановлен после регидратации. Есть основания полагать, что упреждающее начало гемодиализа может улучшить исходы путем удаления нефротоксинов и предотвращения угрожающих жизни осложнений, таких как гиперкалиемия и глубокий метаболический ацидоз. Лечение острой почечной недостаточности следует проводить в соответствии с клиническими рекомендациями, разработанным профессиональными сообществами по нефрологии.
3.2.4 Рекомендации по рациону питания при дефиците SCAD
Так как большинство пациентов с SCADD не имеют симптомов, лечение не требуется, не существует общепринятых рекомендаций по диетотерапии или применению левокарнитина и рибофлавина (биологическая активная добавка).
3.2.5 Рекомендации по рациону питания при дефиците MCAD
Во многих странах проводится неонатальный скрининг на недостаточность MCAD, поэтому пациентов выявляют до начала клинических симптомов. Рекомендации по диетотерапии различаются в зависимости от возраста и статуса подтверждения диагноза.
— Рекомендуется пациентам с установленным диагнозом на недостаточность MCAD прием левокарнитина в дозе 60 — 100 мг на кг в сутки для коррекции вторичного дефицита с целью усиления выведения токсичных метаболитов [130, 149].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Профессиональное сообщество не выработало единых рекомендаций по применению Левокарнитина при MCAD. Некоторые авторы рекомендуют 100 мг/кг/день левокарнитина, чтобы компенсировать вторичный дефицит карнитина и улучшить выведение токсичных метаболитов, другие считают, что в этом нет необходимости [94]. В исследованиях переносимости физических нагрузок у пациентов с дефицитом MCAD было показано, после приема Левокарнитина 100 мг/кг/день переносимость физической нагрузки возрастает [95]. Хотя у пациентов с дефицитом MCAD не было отмечено серьезных побочных эффектов при приеме Левокарнитина, некоторые пациенты жаловались на тошноту, диарею, боль в животе и рыбный запах при приеме в дозе 100 мг/кг/день [96]. Учитывая эту информацию, некоторые врачи рекомендуют использовать низкие дозы Левокарнитина, если уровень свободного карнитина в крови ниже нормы.
3.2.5.1 Рекомендации по диетотерапии новорожденных с MCAD, выявленных при неонатальном скрининге
— Рекомендуются соблюдать интервалы между кормлениями не более 3 — 4 часов для доношенных детей с выявленными изменениями в анализе крови при проведении неонатального скрининга, характерными для дефицита MCAD, пока диагноз не будет подтвержден или исключен на основании дополнительных тестов [98].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Считается, что новорожденные при дефиците MCAD подвергаются наибольшему риску развития гипогликемии в течение первых 72 часов жизни, особенно при грудном вскармливании. Необходимо соблюдать регулярные кормления, не допустимы пропуски ночных кормлений!
— Рекомендуется соблюдать «безопасные» интервалы между кормлением для новорожденных с установленным диагнозом MCAD для предотвращения эпизодов метаболической декомпенсации (Приложение Г6)[98].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Безопасные интервалы между кормлениями были определены на основании опыта стран, проводящих скрининг на дефицит MCAD.
— Рекомендуется новорожденным с установленным диагнозом недостаточность MCAD при грудном вскармливании дополнить питание адаптированной молочной смесью в течение первых трех полных дней (72 часа) для достижения необходимой потребности в калориях [98].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Дети с MCAD, вскармливаемые грудным молоком, подвергаются особому риску, поскольку содержание энергии в грудном молоке в течение первых нескольких дней низкое, и его объемы небольшие. Поэтому рекомендуется дополнить питание адаптированными молочными смесями 60 мл/кг/день. Желательно равномерно разделить объем смеси между 6 и 8 кормлениями и давать после кормления грудью. Если ребенок не усваивает достаточный объем смеси и грудного молока, следует назначить кормление через назогастральный зонд или назначить внутривенную инфузию 10% декстрозой** для достижения необходимой потребности в калориях.
3.2.5.2 Рекомендации по диетотерапии пациентов с MCAD
— Рекомендуется всем пациентам с установленным диагнозом недостаточность MCAD соблюдать интервалы между кормлениями для предотвращения развития гипогликемических состояний (Приложение Г5) [98, 94].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Режим питания у детей старшего возраста и взрослых пациентов должен быть регулярным и включать три основных приема пищи (завтрак, обед, ужин), а также 2 — 3 перекуса (2-ой завтрак, полдник и прием высокоуглеводного продукта перед сном (хлебо-булочные изделия, галеты, крупяное блюдо) с напитком (кисель, компот, чай). Интервал голодания в ночные часы не должен превышать 12 часов.
— Рекомендуется младенцам первого года жизни с установленным диагнозом недостаточность MCAD кормление материнским молоком или молочными смесями без ограничения LCF и без добавок MCT. Рекомендуется соблюдать интервалы между кормлениями для предотвращения развития гипогликемических состояний (Приложение Г5) [98].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Небольшие количества MCT присутствуют в обычных детских смесях, но их количества минимально и не может быть опасным. В высокой концентрации MCT добавляют в некоторые специальные молочные смеси и их не рекомендуют детям с данной формой болезни. Необходимо отказаться от специальных диетических продуктов, содержащих в больших количествах MCT.
Жиры в рационе у пациентов с недостаточностью MCAD не ограничиваются [98].
Среднецепочечные жиры встречаются в некоторых продуктах, например в сливочном масле, коровьем молоке в небольших количествах, и они могут быть включены в рацион без особых ограничений. Кокос является единственным исключением, где 5% жирных кислот имеют длину цепи C8 или C10. Рекомендуют избегать употребления чистого кокоса и кокосового масла, поскольку неизвестно влияние их большого количества на состояние здоровья пациентов с недостаточностью MCAD. MCT добавляются к некоторым специальным смесям для детей и энергетическим добавкам и их не следует включать в рацион питания пациентов. Важно проверить текущий состав всех продуктов на предмет присутствия MCT.
— Рекомендовано у взрослых пациентов при дефиците MCAD избегать длительного голодания для предотвращения гипогликемических состояний и метаболических кризов [94, 98].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Рекомендовано при наличии у пациента с недостаточностью MCAD признаков рекуррентного заболевания, выпаивание растворами, содержащими полимер декстрозы, чтобы предотвратить метаболическую декомпенсацию [94, 98].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Рекомендуют также в период болезни исключать жиры, хотя убедительных доказательств необходимости в этом для недостаточности MCAD не получены. По мере улучшения состояния ребенка нормальная диета может быть возобновлена, но следует давать дополнительные «энергетические» напитки, особенно ночью, до тех пор, пока ребенок полностью не выздоровеет и не перейдет на обычный режим питания.
— Рекомендовано при наличии у пациента с недостаточностью MCAD в период метаболической декомпенсации в случае сложностей приема растворов через рот проведение внутривенной инфузионной терапии растворами, содержащим декстрозу** [94, 98].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Следует начинать инфузионную терапию растворами декстрозы** при одном из следующих показаний: многократная рвота, гипогликемия, низкий PO2, обезвоживание, нарушения сознания, метаболический ацидоз. Подробно описано в разделе 3.1.2 Рекомендации по лечению пациентов с FAOD в период метаболической декомпенсации
— Не рекомендовано пациентам с недостаточностью MCAD в период метаболической декомпенсации введение жиров с целью предотвращения усугубления тяжести состояния [98, 99].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Рекомендуется всем взрослым пациентам с установленным диагнозом недостаточность MCAD избегать диеты с высоким содержанием жиров, употребление алкоголя, что вызывает метаболическую декомпенсацию [94, 99].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Рекомендуется избегать применения ацетилсалициловой кислоты** всем пациентам с установленным диагнозом MCAD с целью предотвращения развития метаболического криза и синдрома Рейе [94, 100].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Ацетилсалициловая кислота** увеличивает митохондриальное окисление жирных кислот, что способствует их накоплению, а также ингибирует пероксисомное окисление. Это приводит к ухудшению состояния пациентов, поскольку окисление жиров в пероксисомах является альтернативным метаболическим путем, позволяющим частично компенсировать биохимический дефект при FAOD.
3.2.6 Рекомендации по рациону питания при дефиците LCHAD и TFP
Дефицит LCHAD — одна из самых тяжелых форм FAOD, при которой наиболее часто развиваются метаболические кризы. Соблюдение диеты рекомендовано всем пациентам в раннем детском возрасте вне зависимости от наличия или отсутствия клинических симптомов [101].
— Рекомендовано у пациентов при дефиците LCHAD и TFP придерживаться максимально низкого потребление LCFA, как при наличии симптомов, так и при их отсутствии, для профилактики возникновения полинейропатии [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Новорожденным показан прием специальной смеси с низким содержанием LCT и высоким содержанием MCT (MCT-содержащая смесь) (Приложение Г10). MCT-содержащая специальная смесь обеспечивает все потребности в питательных веществах. Однако в этом случае необходим дополнительный прием незаменимых длинноцепочечных жирных кислот.
— Рекомендуется назначение диетотерапии при выявлении характерных изменений по результатам неонатального скрининга и/или биохимической диагностики, не дожидаясь подтверждения диагноза молекулярно-генетическим методом [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Поскольку у младенцев может наблюдаться активный липолиз, концентрация некоторых ацилкарнитинов может превышать нормальные значения, что является причиной ложноположительных результатов анализа. В случае если диагноз не будет подтвержден, диетотерапия может быть отменена.
— Рекомендуется при введении твердой пищи в рацион пациентов с недостаточностью LCHAD и MTP поддерживать содержание общих жиров в рационе 25 — 30% от общего потребления калорий, где 20 — 25% приходится на MCT и 5 — 10% — на LCT (Приложение Г11)[66].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Описан положительный эффект повышения потребления белка в отношении энергетического баланса и метаболического контроля у детей с недостаточностью LCHAD или TFP [134].
При дефектах окисления длинноцепочечных жирных кислот следует ограничить потребление длинноцепочечных жиров (вычисляемое как процентная доля суммарной энергии, получаемой из жиров). Согласно рекомендациям D-A-CH (объединение нутриционистов Германии/Австрии/Швейцарии), в рационе здорового младенца доля жиров должна составлять 40 — 45% от общей калорийности, а в рационе ребенка школьного возраста — 30 — 35%. При ограничении длинноцепочечных жирных кислот в рационе необходимы добавки незаменимых жирных кислот для покрытия суточной потребности в жирах (Приложение Г8). Для поддержания оптимального соотношения омега-3 и омега-6 жирных кислот предпочтительны масло грецкого ореха, соевое масло или масло ростков пшеницы [102].
3.2.7 Рекомендации по рациону питания при дефиците VLCAD
В настоящее время не существует научно-обоснованных рекомендаций по ведению пациентов с нарушениями окисления длинноцепочечных жирных кислот [62]. Гетерогенность клинических фенотипов является хорошо установленным фактом [5], что с введением в практику скрининга новорожденных также подразумевает случаи бессимптомного течения болезни [6]. Помимо этого, молекулярная гетерогенность затрудняет прогнозирование степени тяжести заболевания или исхода на момент проведения скрининга новорожденных. Выяснение вопроса о том, необходимо ли при легком фенотипе снижение в рационе доли длинноцепочечных жиров, или достаточно просто ограничиться регулярным кормлением и добавками MCT, особенно при повышенной потребности в калориях, требует более продолжительного наблюдения [66].
В зависимости от состояния ребенка энтеральное питание осуществляется через рот, а также через зонд или гастростому, расчет проводится строго индивидуально.
— Рекомендуется продолжать кормление грудным молоком или адаптированной детской смесью с добавлением специализированной низкожировой смеси с ограничением LCT из расчета 50/50 пациентам с бессимптомным вариантом VLCAD, если результаты стандартных лабораторных анализов, таких как КФК, АЛТ, АСТ и глюкоза в крови находятся в пределах нормы [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Рекомендовано младенцам и более взрослым детям с бессимптомным вариантом VLCAD снижение содержания жиров в рационе до 30 — 40% от общей калорийности из них 10 — 15% энергии за счет MCT с целью предотвращения развития метаболического криза [66].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Некоторые новорожденные с бессимптомным дефицитом VLCAD, у которых профили ацилкарнитинов полностью пришли в норму после первой недели жизни, не получали диету с низким содержанием жиров и модифицированными жирами в первые месяцы жизни, и у них до сих пор не проявились VLCAD-ассоциированные клинические симптомы. Учитывая молекулярную гетерогенность при VLCADD, очень трудно установить четкую корреляцию генотип-фенотип и спрогнозировать клиническое течение болезни. Однако мутация p.V243A, как минимум, одна копия которой обнаруживается у многих бессимптомных пациентов, выявляемых при скрининге новорожденных, как правило, ассоциирована с мягким фенотипом. У пациентов с этой мутацией может развиваться гипогликемия при тяжелых нарушениях здоровья или миопатия при сильной физической нагрузке, но возможно и бессимптомное течение.
Для пациентов, у которых развиваются только связанные с физической нагрузкой миопатические симптомы в более позднем возрасте, обогащение рациона MCT (особенно перед физической нагрузкой) может играть важную роль, а необходимость в ограничении LCT может отсутствовать.
— Рекомендуется при наличии симптомов у пациентов с дефицитом VLCAD диета с пониженным содержанием LCFA и перевод пациента на модифицированные жиры (добавки MCT) для предотвращения мышечной боли и рабдомиолиза [61, 66].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: при наличии симптомов содержание общих жиров 25 — 30% от общей калорийности, из них 20 — 25% за счет MCT и 5 — 10% за счет LCT (Приложение Г12)[61].
Поскольку клинические симптомы при долгосрочном наблюдении у пациентов с дефицитом VLCAD представлены преимущественно эпизодической мышечной болью и рабдомиолизом, что указывает на недостаток мышечной энергии, рацион необходимо обогащать MCT, которые должны составлять 20% от общей калорийности (Таблица 3). Пациентам с мышечной слабостью и болью при физических нагрузках помогает повышение потребления MCT (или углеводов) непосредственно перед более интенсивной физической нагрузкой (напр., в дозировке 0,25 — 0,5 г MCT/кг).
3.2.8 Рекомендации по лечению пациентов с ГА2
— Рекомендуется пациентам с установленным диагнозом ГА2 соблюдение диеты с низким содержанием жиров (20 — 25% энергии), низким содержанием белка для снижения избыточного потребления изолейцина, лейцина, лизина, триптофана и валина [103, 111].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Не рекомендуется пациентам с установленным диагнозом ГА2 назначение MCT [104].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Рекомендуется во время кризов пациентам с установленным диагнозом ГА2 парентеральное введение декстрозы** для купирования гипогликемии и подавления липолизиса, бензоата натрия (биологически активная добавка) — для купирования гипераммониемии [111].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: детальное описание приведено в 3.1.2 Рекомендации по лечению пациентов с FAOD в период метаболической декомпенсации
3.3 Рекомендации по применению отдельных лекарственных препаратов
Кроме применения диетотерапии при нарушениях окисления жирных кислот также необходимо применение ряда лекарственных препаратов и витаминов, чтобы компенсировать дефицит незаменимых жирных кислот в организме, карнитина и жирорастворимых витаминов.
3.3.1 Рекомендации по применению отдельных лекарственных препаратов, содержащих левокарнитин
— Рекомендуется избегать назначения Левокарнитина в период криза при дефектах окисления длинноцепочечных жирных кислот, особенно в виде внутривенных инфузий для предотвращения развития рабдомиолиза, неврологических нарушений и аритмии [63, 142, 132].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Левокарнитин является основной терапией пациентов при первичной недостаточности карнитина. Также при низких значения карнитина в крови может использоваться при других формах FAOD с целью усиления связывания метаболитов жирных кислот и выведения их с мочой. Однако использование левокарнитина при FAOD является спорным, т.к. доказательства его эффективности при вторичном дефиците карнитина отсутствуют. При недостаточности MTP и LCHAD добавки с Левокарнитином могут индуцировать дальнейшую продукцию 3-гидроксиацилкарнитинов, способных вызывать необратимые невропатические симптомы и аритмию. Исследования на мышах с моделью дефицита VLCAD также показали, что нехватка карнитина вызывает его эндогенный биосинтез), который, однако, подавляется при экзогенном введении Левокарнитина. У многих пациентов с дефектами окисления жирных кислот, не получающих добавок Левокарнитина, концентрации свободного карнитина в плазме также варьируют от низких до нормальных, что позволяет говорить и об эндогенном биосинтезе карнитина у человека. Таким образом, добавки, содержащие Левокарнитин, должны назначаться с осторожностью, особенно, когда речь идет о недостаточности TFP и LCHAD. Существует консенсусное мнение, согласно которому следует отказаться, в частности, от внутривенного введения Левокарнитина при острых метаболических нарушениях, поскольку в таких ситуациях повышается продукция потенциально токсичных ацилкарнитинов, что приводило к внезапной смерти пациентов.
3.3.2 Рекомендации по применению незаменимых жирных кислот
Пациенты с FAOD, находящиеся на низкожировой диете подвержены высокому риску дефицита жирорастворимых витаминов. Важно избегать дефицита незаменимых жирных кислот, и большая часть потребления длинноцепочечных жиров должна поступать из масел, богатых незаменимыми жирными кислотами. Для удовлетворения потребностей в незаменимых жирных кислотах может потребоваться добавление специальных масел, таких как ореховое или льняное масло (Приложение Г13)[105].
— Рекомендуется пациентам с недостаточностью MTP и LCHAD, назначать биологическую активную добавку в виде докозагексаеновой кислоты (DHA), играющей важную роль в формировании функций нервной и иммунной систем, а также зрительного анализатора, в дозировке 60 — 65 мг/день детям с весом тела < 20 кг и в дозировке 130 мг в день — детям с весом тела > 20 кг для поддержания функций нервной и иммунной системы [105, 137, 157].
Уровень убедительности доказательств B (уровень достоверности рекомендации — 3).
Комментарии: Линолевая кислота и -линоленовая кислота являются предшественниками для эндогенного синтеза более длинных жирных кислот, таких как эйкозапентаеновая кислота (EPA) и докозагексаеновая кислота (DHA), которые имеют решающее значение для формирования функций нервной, иммунной систем и зрительного анализатора [105]. Обеспечить оптимальную концентрацию этих жирных кислот в плазме только с помощью применения пищевых масел сложно, поэтому пациентам требуется добавление их в чистом виде для достижения нормального уровня. Считается, что добавки DHA могут стабилизировать, и затормозить развитие пигментной дегенерации сетчатки при FAOD. Согласно проведенному исследованию [157], оптимальная диетотерапия, назначаемая на основании низких концентраций 3-гидроксиацилкарнитина и высоких концентраций DHA в плазме, позволяла сохранять функцию сетчатки и остроту зрения у детей с недостаточностью LCHAD или MTP.
— В межприступный период для пациентов с FAOD находящихся на диете с ограничением длинноцепочечных жиров рекомендовано добавлять натуральные растительные масла (кокосовое масло, масло грецкого ореха) для восполнения недостатка жирных кислот [105].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Соотношение незаменимых жирных кислот в различных маслах приведено в Приложении Г13.
— Рекомендовано назначение незаменимых жирных кислот пациентам с FAOD на фоне низкожировой диетотерапии с целью компенсации их недостаточности [11 — 14, 105].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: В приложении Г8 приведены основные рекомендации по назначению незаменимых жирных кислот при диете с низким содержанием жиров.
3.3.3 Рекомендации по применению среднецепочечных триглицеридов
— Рекомендовано в качестве основных пищевых источников среднецепочечных жирных кислот использовать 50% эмульсию MCT в питании пациентов с недостаточности TFP и LCHAD и VLCAD для предотвращения развития мышечной симптоматики [112, 149].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Содержание MCT в различных продуктах лечебного питания приведено в приложении Г14. Эпизодические мышечные симптомы, например, рабдомиолиз, патогенетически связывают с дефицитом энергии, и их можно преодолеть с помощью углеводов или MCT. В частности, по имеющимся данным, связанный с физической активностью рабдомиолиз можно предотвратить путем приема достаточного количества MCT непосредственно перед нагрузкой
— Не рекомендуется назначение пациентам с MCAD триглицеридов со средней длиной цепи (MCT) с целью предотвращения ухудшения состояния пациента, развития метаболического криза [105].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Жирные кислоты со средней длиной цепи могут проникать в митохондрии, не будучи связанными с карнитином, минуя этап, на котором обычно регулируется окисление жирных кислот. Поэтому прием MCT может привести к более серьезным нарушениям обмена веществ, чем при использовании длинноцепочечных жиров.
3.3.4 Рекомендации по применению витаминов
— Рекомендуется пациентам с FAOD назначение витаминов группы B и жирорастворимых витаминов A, D, E в возрастных дозировках с целью предотвращения их дефицита [5, 12, 105].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Витамины назначают в возрастных профилактических дозах курсами по 2 — 3 месяца.
— Рекомендуется всем пациентам с биохимическими нарушениями, характерными для ГА2 назначение (биологической активной добавки) рибофлавина [106, 111, 135, 136].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Сходный метаболический профиль может наблюдаться при дефекте транспортера рибофлавина, поэтому назначение рибофлавина (биологическая активная добавка) per os в дозе 100 — 200 г/день позволяет полностью купировать клинические и биохимические симптомы болезни.
3.4 Симптоматическое лечение
— Не рекомендуется применять препараты вальпроевой кислоты** в случае необходимости назначения противоэпилептической терапии с целью предотвращения развития печеночной недостаточности [5, 12, 145].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: У пациентов с дефектами митохондриального -окисления жирных кислот применение вальпроевой кислоты** может привести к развитию тяжелой печеночной недостаточности.
3.5 Хирургическое лечение
При возникновении показаний для хирургических вмешательств рекомендуется коллегиальное принятие решения о тактике его проведения [5, 12].
4. Медицинская реабилитация, медицинские показания и противопоказания к применению методов реабилитации
Пациентам с врачом-физиотерапевтом и врачом по лечебной физкультуре разрабатывается персонализированный курс реабилитации, включающий массаж, лечебную физкультуру, физиотерапевтические процедуры (магнитотерапию, термотерапию, ударно-волновую терапию, метод биологической обратной связи и другие процедуры).
Реабилитационные курсы (массаж, ЛФК, физиопроцедуры, психолого-педагогическая помощь) рекомендуется проводить в условиях дневного стационара с частотой 3 — 4 раза в год [1, 2, 14].
— Рекомендуется пациентам и членам их семей проводить консультацию/консультации медицинского психолога с целью повышения приверженности к лечению [1, 2, 14].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: Основной целью работы паллиативных служб является создание всех необходимых условий для обеспечения нахождения пациентов в домашних условиях, а не в стенах лечебного учреждения, что позволяет не только улучшить качество жизни пациентов и их семей, но и существенно снизить государственные затраты на постоянное стационарное лечение таких пациентов [1, 2, 14].
5. Профилактика и диспансерное наблюдение, медицинские показания и противопоказания к применению методов профилактики
5.1 Профилактика
Медико-генетическое консультирование
— Рекомендуется медико-генетическое консультирование после установления диагноза пациентам с FAOD, с целью разъяснений генетического риска, возможности проведения пренатальной и преимплантационной диагностики для любой последующей беременности в семьях, отягощенных хотя бы одним случаем FAOD [1, 2, 6].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарий: Семьям с больными детьми рекомендуется медико-генетическое консультирование с целью определения генетического риска. Как и при других аутосомно-рецессивных заболеваниях, для каждой беременности риск рождения ребенка составляет 25%. В семьях, где есть больной ребенок, возможно проведение пренатальной и преимплантационной диагностики.
Пренатальная диагностика проводится молекулярно-генетическими методами, путем исследования ДНК, выделенной из биоптата ворсин хориона на 9 — 11 неделе беременности и/или клеток амниотической жидкости, плодной крови на 20 — 22 неделе беременности. При мягких формах FAOD хорошо отвечающих на терапию, решение о проведении пренатальной диагностики должно быть принято только после подробного обсуждения с семьей всех рисков и прогноза заболевания у потомства, возможностей лечения при ранней диагностике.
Здоровый образ жизни
Подростки и взрослые с FAOD должны быть осведомлены о рисках, связанных с чрезмерным употреблением алкоголя. Алкоголь ингибирует глюконеогенез, поэтому нарушаются как окисление жирных кислот, так и глюконеогенез, которые обеспечивают организм метаболической энергией во время голодания. Необходимо рекомендовать ограничение потребления алкоголя и всегда в сочетании с едой.
Детей с установленным диагнозом FAOD следует учить «здоровому образу жизни» с помощью диеты и физических упражнений. Некоторые родители обеспокоены тем, что их ребенок не ест достаточно. Если ребенок не съедает рекомендованный объем, порцию, при необходимости можно докормить или организовать перекус позже, чтобы обеспечит физиологическую потребность в энергии.
Люди с дефицитом MCAD не нуждаются в дополнительных калориях; следует избегать переедания из-за риска ожирения.
Комментарии: Долгосрочные результаты исследований показали, что люди, лечившиеся от дефицита MCAD, склонны к чрезмерному увеличению веса [108]. Дети в препубертатном возрасте могут иметь избыточный вес, вследствие частого кормления как часть лечения. Показатели роста и массы тела должны тщательно контролироваться при каждом посещении клиники. Динамическое наблюдение должно включать рекомендации по контролю массы тела, обучение здоровому образу жизни, включающему в себя дробное регулярное питание с соблюдением норм физиологической калорийности и рекомендации по физической активности.
Наблюдение в период беременности
— Рекомендуется консультация женщин-носительниц FAOD для разъяснения рисков возникновения акушерских осложнений [105, 138].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5)
Комментарий: Женщины, у которых был ребенок с FAOD подвержены риску во время последующих беременностей HELLP синдрому. Особенно это касается следующих заболеваний: дефициты LCHAD, VLCAD, CPT1, CPT2 и CACT.
5.2 Диспансерное наблюдение
— Рекомендуется определение концентрации ацилкарнитинов в крови у пациентов с FAOD с целью контроля уровня свободного карнитина и ацилкарнитинов [19, 21].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 4)
Комментарии: Определение ацилкарнитинов проводится с частотой: каждые 1 — 2 месяца пациентам до года, каждые 3 — 6 месяцев у детей младше 6 лет, каждые 6 — 12 месяцев пациентам старше 6 лет, по показаниям в период и при подозрении на метаболический криз. Необходимо отметить, что уровень ацилкарнитинов может повышаться при назначении Левокарнитина (Приложение Г16).
— Рекомендовано всем пациентам с диагнозом FAOD общий (клинический) анализ крови развернутый (гемоглобин, количество эритроцитов, цветовой показатель, количество лейкоцитов, тромбоцитов, лейкоцитарная формула и скорость оседания эритроцитов) для оценки основных параметров кроветворения и наличия воспалительных процессов [1, 2, 5, 28].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: в связи с высоким риском развития интеркуррентных инфекций общий анализ крови пациентам с FAOD проводится с частотой: каждые 6 месяцев пациентам до 6 лет, каждые 6 — 12 месяцев пациентам старше 6 лет.
— Рекомендуется всем пациентам с FAOD проведение анализа крови биохимического общетерапевтического (содержание глюкозы, общего белка, белковых фракций, альбумин, C-реактивного белка, общего билирубина и его фракции, холестерина, триглицеридов, липопротеидов низкой и высокой плотности (исследование уровня липопротеинов в крови), щелочной фосфатазы, креатинина, мочевины, мочевой кислоты, аммония, аланинаминотрансферазы (АЛТ), аспартатаминотрансферазы (АСТ), гаммаглютамилтрансферазы (ГГТ), креатинфосфокиназы (КФК), Исследование уровня/активности изоферментов креатинкиназы в крови, лактатдегидрогеназа (ЛДГ), кальция общего и ионизированного, натрия, калия, неорганического фосфора, железа, ферритина, магния, хлора, молочной кислоты), инсулина, показателей кислотно-основного равновесия (Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (pH) крови), исследование уровня молочной кислоты в крови (лактата) с целью оценки состояния печени, почек, баланса важнейших нутриентов при диетотерапии, кальциево-фосфорного обмена и выявления отклонений важных биохимических показателей для дальнейшей коррекции [12, 132, 145].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5)
Комментарий: анализ проводится не реже 1 раза в 6 месяцев и по показаниям при ухудшении состояния и при метаболическом кризе.
— Рекомендовано всем пациентам с FAOD общий (клинический) анализ мочи, определение кетоновых тел для оценки состояния мочевыводящих путей и почек [1, 2, 5, 34].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
Комментарии: в связи с высоким риском развития интеркуррентных инфекций у пациентов с FAOD рекомендуется проведение данного исследования не реже 5 раз в год
Обычно общий анализ мочи пациентам с FAOD проводится с частотой: каждые 6 месяцев пациентам до 6 лет, каждые 6 — 12 месяцев пациентам старше 6 лет, чаще — по показаниям при ухудшении состояния и при метаболическом кризе.
— Рекомендуется определение лабораторных показателей (кислотно-щелочного состояния крови (Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (pH) крови), исследование уровня молочной кислоты в крови (лактата), глюкоза, электролиты (калий, натрий, кальций ионизированный, хлор) в период и при подозрении на метаболический криз пациентам с клиническими и/или биохимическими признаками FAOD с целью оценки состояния и своевременной коррекции терапии [12, 132].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 5).
— Рекомендуется всем пациентам с патологией сердечно-сосудистой системы исследования уровня N-терминального фрагмента натрийуретического пропептида мозгового (NT-proBNP) в крови для своевременной диагностики сердечной недостаточности, дифференциальной диагностики с одышкой, вызванной респираторными нарушениями, для решения вопросов о старте/коррекции кардиотропной терапии [11, 133].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 4).
— Рекомендуется проведение холтеровского мониторирования ЭКГ, Эхо-КГ, пациентам с FAOD при постановке диагноза, далее не реже 1 раза в год с целью диагностики патологии со стороны ССС [97, 109, 129, 133].
Уровень убедительности доказательств C (уровень достоверности рекомендации — 4).
— Рекомендуется проведение электроэнцефалографии (ЭЭГ) пациентам с FAOD при наличии неврологической симптоматики для выявления эпилептической активности и для контроля противоэпилептической терапии согласно клиническим рекомендациям по эпилепсии [35, 158].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 4)
Комментарии: в том числе ЭЭГ с видеомониторингом. При отсутствии приступов — не реже 1 раза в год, при наличии эпилептических приступов — по показаниям.
— Рекомендуется проведение ультразвукового исследования (УЗИ) органов брюшной полости пациентам с FAOD для выявления патологии печени и почек [9, 12, 13, 145].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5)
Комментарии: Кратность при отсутствии изменений — не реже 1 раза в год, при наличии патологии печени — по показаниям.
— Рекомендуется проведение офтальмоскопии пациентам с FAOD с целью своевременного обнаружения офтальмологической патологии [38, 149].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5)
Комментарии: Кратность при отсутствии изменений — не реже 1 раза в год, при наличии патологии — по показаниям.
— Рекомендуется пациентам с FAOD проведение коррекции лечебного питания и симптоматической терапии с целью исключения белково-энергетической недостаточности [11 — 14, 105].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5)
Комментарии: Коррекцию диетотерапии проводят врачи-генетики или врачи-диетологи с частотой 1 р/мес. на 1 году жизни, далее 1 р/3 мес. и по показаниям до 3 лет; 1 р/6 мес. после 3 лет.
— Рекомендуются для наблюдения пациентов с установленным диагнозом FAOD применять мультидисциплинарный подход ввиду того, что заболевания характеризуются поражением многих органов и систем, требует комплексной терапии, что диктует необходимость совместного ведения пациента специалистами разных профилей [12, 13, 15, 28, 145].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5)
Комментарии: показаны первичные и повторные консультации врача-генетика, врача-офтальмолога, врача-невролога, врача-гастроэнтеролога, врача-эндокринолога, врача-кардиолога, врача-детского кардиолога, врача-нефролога, врача-педиатра/врача-терапевта/врача общей практики (семейного врача), а также врачей других специальностей пациентам с FAOD, имеющим нарушения функций соответствующих органов и систем
— Рекомендуется обучение родителей (законных представителей)/пациентов правилам организации терапии в межприступный период и в период угрозы метаболического криза пациентов с FAOD с целью предотвращения развития повторных метаболических кризов [105].
Уровень убедительности рекомендаций C (уровень достоверности доказательств — 5)
Комментарии: У родителей ребенка всегда должна быть памятка с указанием неотложных мероприятий в период начинающегося метаболического криза (Приложение Г17).
6. Организация оказания медицинской помощи
Показания для плановой госпитализации:
— необходимость проведения различных видов экспертиз или обследования в медицинской организации при невозможности проведения их в амбулаторных условиях, требующих динамического наблюдения (в том числе оформление заключения федерального консилиума).
Показания для экстренной госпитализации:
— острые заболевания;
— обострения хронических болезней;
— отравления и травмы, состояния, требующие интенсивной терапии и перевода в реанимационные отделения или отделения интенсивной терапии а также круглосуточного медицинского наблюдения и проведения специальных видов обследования и лечения.
Показания к выписке пациента из медицинской организации:
— отсутствие угрозы жизни больного;
— отсутствие угрозы развития осложнений, требующих неотложного лечения;
— стабилизация состояния и основных клинико-лабораторных показателей патологического процесса по основному заболеванию;
— отсутствие необходимости в постоянном врачебном и круглосуточном медицинском наблюдении по основному заболеванию;
— необходимости перевода больного в другую больницу или учреждение социального обеспечения.
Пациентам с FAOD 1 раз в 6 — 12 мес. (в соответствии с тяжестью состояния) показано комплексное обследование в многопрофильных стационарах.
Продолжительность госпитализации зависит от скорости коррекции метаболических нарушений, а также от сроков появления положительной динамики со стороны центральной нервной системы и других органов, скорости восстановления показателей глюкозы крови и кислотно-щелочного состояния, ответ на лечение отмечается в течение 5 — 7 дней. Пребывание в стационаре в среднем составляет 21 день.
После выписки из стационара дети нуждаются в наблюдении педиатра, невролога, кардиолога, гастроэнтеролога, офтальмолога, диетолога, генетика, проведении ЭКГ (по показаниям).
Наблюдение пациентов по месту жительства (в амбулаторно-поликлинических условиях) должно осуществляться постоянно.
7. Дополнительная информация (в том числе факторы, влияющие на исход заболевания или состояния)
Продолжительность жизни во многом зависит от тяжести заболевания и проведения патогенетической и симптоматической терапии. Причиной смерти чаще всего являются сердечно-легочная недостаточность.
В случае показания к диетотерапии, следует начать лечение как можно раньше с целью предотвращения прогрессирования осложнений [1 — 4, 11 — 14].
Прогноз состояния и уровня психического развития пациентов зависит от тяжести заболевания, степени поражения внутренних органов (сердце, печень) и ЦНС, сроков начала лечения и эффективности интенсивной терапии при метаболической декомпенсации. Рано манифестирующая системная форма заболевания обычно имеет более тяжелое течение и менее благоприятный прогноз.
Неблагоприятные лабораторные признаки, свидетельствующие о несбалансированности терапии и угрозе развития метаболического криза: уменьшение содержания свободного карнитина, глюкозы, тенденция к снижению pH крови, дефициту оснований, нарастание содержания ацилкарнитинов с очень длинной цепью (при снижении показателя свободного карнитина) в крови и повышение экскреции органических кислот.
На недостаточность нутритивной поддержки указывает снижение уровня гемоглобина, железа, общего белка и альбумина. Низкий уровень в крови незаменимых жирных кислот (линолевой, линоленовой, арахидоновой, докозагексаеновой и др.), нарушенное соотношение линолевой/линоленовой кислот (в норме 5:1 — 10:1) могут обусловить задержку психомоторного и речевого развития, созревание иммунной системы и снижение остроты зрения.
Одним из эффективных подходов к ранней диагностике FAOD является массовый скрининг новорожденных. Неонатальный скрининг в Российской Федерации на FAOD проводится в отдельных регионах [1 — 3].
Все должны иметь часто обновляемое «экстренное» письмо, которое может при необходимости передаваться медицинским работникам, которые могут не знать о FAOD Пример такой памятки приведен в Приложении Г17.
Критерии оценки качества медицинской помощи
N | Критерии качества | Оценка выполнения |
1. | Выполнено определение концентрации ацилкарнитинов (при постановке диагноза) | Да/нет |
2. | Выполнено назначение биохимического анализа крови общетерапевтического (АСТ, АЛТ, КФК, глюкоза, ЛДГ, холестерин, триглицериды) всем пациентам с клиническими и/или биохимическими признаками FAOD (при постановке диагноза и далее каждые 6 месяцев) | Да/нет |
3. | Выполнено исследование кислотно-основного состояния и газов крови при постановке диагноза и при угрозе метаболического криза | Да/нет |
4. | Выполнено молекулярно-генетическое исследование при постановке диагноза | Да/нет |
5. | Выполнена ЭКГ, эхокардиография всем пациентам с клиническими и/или биохимическими признаками FAOD (при постановке диагноза и далее не реже 1 раза в год) | Да/нет |
6. | Выполнено определение размеров печени и селезенки по данным ультразвукового исследования всем пациентам с клиническими и/или биохимическими признаками FAOD (при постановке диагноза и далее при отсутствии изменений — не реже 1 раза в год, при наличии патологии печени — по показаниям) | Да/нет |
7. | Выполнено электроэнцефалография (при отсутствии приступов — 1 ргод) | Да/нет |
8. | Выполнена консультация врачом-генетиком при установлении диагноза | Да/нет |
9. | Выполнена консультация врачом-кардиологом/врачом-детским кардиологом при наличии патологии сердечно-сосудистой системы | Да/нет |
10. | Выполнена консультация врачом-неврологом при наличии патологии со стороны центральной нервной системы | Да/нет |
11. | Выполнена консультация врачом-офтальмологом (при наличии патологии зрения) | Да/нет |
12. | Выполнена консультация врачом-педиатром/врачом-терапевтом не реже 1 раза в 6 месяцев | Да/нет |
13. | Выполнена консультация врачом-диетологом (при постановке диагноза и далее с частотой 1 р/мес. на 1 году жизни, далее 1 р/3 мес. и по показаниям до 3 лет; 1 р 6 мес. после 3 лет) с перерасчетом диеты в зависимости от прибавки массы тела и возраста | Да/нет |
Список литературы
1. Михайлова С.В., Захарова Е.Ю., Петрухин А.С. Нейрометаболические заболевания у детей и подростков: диагностика и подходы к лечению (2-е изд., переработанное и дополненное) / М.: Литтерра, 2022.
2. Orngreen MC, Norgaard MG, Sacchetti M, van Engelen BG, Vissing J. Fuel utilization in patients with very long-chain acyl-coa dehydrogenase deficiency. Ann Neurol. 2004 Aug; 56(2): 279 — 83
3. Краснопольская К.Д. Наследственные болезни обмена веществ. Справочное пособие для врачей. / М., 2005. С. 43 — 51.
4. Andresen BS, Olpin S, Poorthuis BJ et al (1999) Clear correlation of genotype with disease phenotype In very-long-chain acyl-CoA dehydrogenase deficiency. Am J Hum Genet 64: 479 — 494;
5. Gregersen N, Andresen BS, Corydon MJ, Corydon TJ, Olsen RK, Bolund L, Bross P. Mutation analysis in mitochondrial fatty acid oxidation defects: Exemplified by acyl-CoA dehydrogenase deficiencies, with special focus on genotype-phenotype relationship. Hum Mutat. 2001 Sep; 18(3): 169 — 89;
6. Spiekerkoetter U, Khuchua Z, Yue Z, Bennett MJ, Strauss AW. General mitochondrial trifunctional protein (TFP) deficiency as a result of either alpha- or beta-subunit mutations exhibits similar phenotypes because mutations in either subunit alter TFP complex expression and subunit turnover. Pediatric Research. 2004; 55(2): 190 — 196. doi: 10.1203/01.PDR.0000103931.80055.06
7. Wilcken B.Recent advances in newborn screening. J Inherit Metab Dis. 2007 Apr; 30(2): 129 — 33.
8. Wilcken B. Fatty acid oxidation disorders: outcome and long-term prognosis. J Inherit Metab Dis. 2022; 33(5): 501 — 506
9. Baruteau J, Sachs P, Broue P, Brivet M, Abdoul H, Vianey-Saban C, Ogier de Baulny H. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2022 Sep; 36(5): 795 — 803
10. Saudubray JM, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999; 22(4): 488 — 502.
11. Bonnet D, et al. Arrhythmias and conduction defects as presenting symptoms of fatty acid oxidation disorders in children. Circulation. 1999; 100(22): 2248 — 2253.
12. Wanders R.J.A., Vreken P., den Boer M.E.J. et al. Disorders of mitochondrial fatty acyl-CoA -oxidation. J Inher Metab Dis 1999; 22: 442 — 487.
13. Николаева Е.А., Мамедов И.С. «Диагностика наследственных дефектов обмена жирных кислот у детей» Российский вестник перинатологии и педиатрии, 2008: 6: 66 — 80
14. Николаева Е.А., Леонтьева И.В., Калачанова Е.П., Золкина И.В. Задержка физического развития и кардиомиопатия у ребенка с первичным системным дефицитом карнитина. Трудный пациент 2022; 2 — 3: 50 — 54.
15. МР 2.3.1.2432-08 «Нормы физиологических потребностей в энергии и пищевых веществах для различных групп населения Российской Федерации» (утв. Главным государственным санитарным врачом РФ 18 декабря 2008 г.).
16. Wilcken B, et al. Pregnancy and fetal long-chain 3-hydroxyacyl coenzyme a dehydrogenase deficiency. Lancet. 1993; 341(8842): 407 — 408
17. Makhseed N, et al. Carnitine transporter defect due to a novel mutation in the SLC22A5 gene presenting with peripheral neuropathy. J Inherit Metab Dis. 2004; 27(6): 778 — 780
18. Wang Y, et al. Phenotype and genotype variation in primary carnitine deficiency. Genet Med. 2001; 3(6): 387 — 392.
19. Innes AM, et al. Hepatic carnitine palmitoyltransferase I deficiency presenting as maternal illness in pregnancy. Pediatr Res. 2000; 47(1): 43 — 45
20. Al-Thihli K, Sinclair G, Sirrs S, Mezei M, Nelson J, Vallance H. Performance of serum and dried blood spot acylcarnitine profiles for detection of fatty acid -oxidation disorders in adult patients with rhabdomyolysis. J Inherit Metab Dis. 2022. Mar; 37(2): 207 — 13.
21. Arnold GL, et al. A Delphi clinical practice protocol for the management of very long chain acyl-CoA dehydrogenase deficiency. Mol Genet Metab. 2009; 96(3): 85 — 90
22. Bach AC, Babayan VK. Medium-chain triglycerides: an update. Am J Clin Nutr. 1982; 36(5): 950 — 962. doi: 10.1093/ajcn/36.5.950.
23. Bartlett K, Eaton S. Mitochondrial -oxidation. Eur J Biochem. 2004; 271(3): 462 — 469.
24. Baruteau J, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study of 187 patients. J Inherit Metab Dis. 2022; 36(5): 795 — 803.
25. Baruteau J, et al. Clinical and biological features at diagnosis in mitochondrial fatty acid beta-oxidation defects: a French pediatric study from 187 patients. Complementary data. J Inherit Metab Dis. 2022; 37(1): 137 — 139.
26. Bastin J, Lopes-Costa A, Djouadi F. Exposure to resveratrol triggers pharmacological correction of fatty acid utilization in human fatty acid oxidation-deficient fibroblasts. Hum Mol Genet. 2022; 20(10): 2048 — 2057.
27. Berardo A, DiMauro S, Hirano M. A diagnostic algorithm for metabolic myopathies. Curr Neurol Neurosci Rep. 2022; 10: 118 — 26.
28. Bleeker JC, Houtkooper RH. Sirtuin activation as a therapeutic approach against inborn errors of metabolism. J Inherit Metab Dis. 2022; 39: 565 — 572.
29. Bleeker JC, Kok IL, Ferdinandusse S, et al. Proposal for an individualized dietary strategy in patients with very long-chain acyl-CoA dehydrogenase deficiency. J Inherit Metab Dis 2022.
30. Bonnefont JP, et al. Long-term follow-up of bezafibrate treatment in patients with the myopathic form of carnitine palmitoyltransferase 2 deficiency. Clin Pharmacol Ther. 2022; 88(1): 101 — 108.
31. Borch L, et al. Normal levels of plasma free carnitine and Acylcarnitines in follow-up samples from a Presymptomatic case of carnitine Palmitoyl transferase 1 (CPT1) deficiency detected through newborn screening in Denmark. JIMD Rep. 2022; 3: 11 — 15.
32. Burrage LC, et al. Elevations of C14: 1 and C14: 2 plasma Acylcarnitines in fasted children: a diagnostic dilemma. J Pediatr. 2022; 169: 208 — 213. e2.
33. Chegary M, et al. Mitochondrial long chain fatty acid beta-oxidation in man and mouse. Biochim Biophys Acta. 2009; 1791(8): 806 — 815.
34. Cox GF, et al. Reversal of severe hypertrophic cardiomyopathy and excellent neuropsychologic outcome in very-long-chain acyl-coenzyme a dehydrogenase deficiency. J Pediatr. 1998; 133(2): 247 — 253.
35. Cox PJ, et al. Nutritional ketosis alters fuel preference and thereby endurance performance in athletes. Cell Metab. 2022; 24(2): 256 — 268.
36. De Biase I, et al. Diagnosis, treatment, and clinical outcome of patients with mitochondrial trifunctional protein/long-chain 3-Hydroxy acyl-CoA dehydrogenase deficiency. JIMD Rep. 2022; 31: 63 — 71. doi: 10.1007/8904_2022_558.
37. Demaugre F, et al. Hepatic and muscular presentations of carnitine palmitoyl transferase deficiency: two distinct entities. Pediatr Res. 1988; 24(3): 308 — 311.
38. Deschauer M, Wieser T, Zierz S. Muscle carnitine palmitoyltransferase II deficiency: Clinical and molecular genetic features and diagnostic aspects. Arch Neurol. 2005; 62: 37 — 41.
39. Diekman E, et al. The newborn screening paradox: sensitivity vs. Overdiagnosis in VLCAD deficiency. JIMD Rep. 2022; 27: 101 — 106.
40. Diekman EF, Ferdinandusse S, van der Pol L, Waterham HR, Ruiter JP, Ijlst L, Wanders RJ, Houten SM, Wijburg FA, Blank AC, Asselbergs FW, Houtkooper RH, Visser G. Fatty acid oxidation flux predicts the clinical severity of VLCAD deficiency. Genet Med. 2022 Apr 2.
41. Diekman EF, van Weeghel M, Wanders RJ, Visser G, Houten SM. Food withdrawal lowers energy expenditure and induces inactivity in long-chain fatty acid oxidation-deficient mouse models. FASEB J. 2022 Jul; 28(7): 2891 — 900.
42. Djouadi F, et al. Bezafibrate increases very-long-chain acyl-CoA dehydrogenase protein and mRNA expression in deficient fibroblasts and is a potential therapy for fatty acid oxidation disorders. Hum Mol Genet. 2005; 14(18): 2695 — 2703.
43. Djouadi F, et al. Peroxisome proliferator activated receptor delta (PPARdelta) agonist but not PPARalpha corrects carnitine palmitoyl transferase 2 deficiency in human muscle cells. J Clin Endocrinol Metab. 2005; 90(3): 1791 — 1797.
44. Elsayed EF, Reilly RF. Rhabdomyolysis: A review, with emphasis on the pediatric population. Pediatr Nephrol. 2022; 25: 7 — 18.
45. Erguven Muferet, Yilmaz Oznur, Koc Seher, Caki Suar, Ayhan Yusuf, Donmez Metin, Dolunay Gulderen. A Case of Early Diagnosed Carnitine Deficiency Presenting with Respiratory Symptoms. Annals of Nutrition and Metabolism. 2007; 51(4): 331 — 334. doi: 10.1159/000107675.
46. Evans M, et al. VLCAD deficiency: follow-up and outcome of patients diagnosed through newborn screening in Victoria. Mol Genet Metab. 2022; 118(4): 282 — 287.
47. Gillingham MB, et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: a double blinded, randomized controlled trial. J Inherit Metab Dis. 2022; 40(6): 831 — 843.
48. Houten SM, Violante S, Ventura FV, Wanders RJA. The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Annu Rev Physiol. 2022; 78: 23 — 44.
49. Ibdah JA. Acute fatty liver of pregnancy: an update on pathogenesis and clinical implications. World J Gastroenterol. 2006; 12(46): 7397 — 7404.
50. IJlst L, et al. Common missense mutation G1528C in long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. Characterization and expression of the mutant protein, mutation analysis on genomic DNA and chromosomal localization of the mitochondrial trifunctional protein alpha subunit gene. J Clin Invest. 1996; 98(4): 1028 — 1033.
51. Kilfoyle D, Hutchinson D, Potter H, George P. Recurrent myoglobinuria due to carnitine palmitoyltransferase II deficiency: Clinical, biochemical, and genetic features of adult-onset cases. N Z Med J. 2005; 118: U1320.
52. Kluge Stefan, Kuhnelt Peter, Block Andreas, Merkel Martin, Gocht Andreas, Lukacs Zoltan, Kohlschutter Alfried, Kreymann Georg. A young woman with persistent hypoglycemia, rhabdomyolysis, and coma: Recognizing fatty acid oxidation defects in adults. Critical Care Medicine. 2003; 31(4): 1273 — 1276.
53. Liu J, Ghaziani TT, Wolf JL. Acute fatty liver disease of pregnancy: updates in pathogenesis, diagnosis, and management. Am J Gastroenterol. 2022; 112(6): 838 — 846.
54. Longo N, Amat di San Filippo C, Pasquali M. Disorders of carnitine transport and the carnitine cycle. Am J Med Genet C Semin Med Genet. 2006; 142c(2): 77 — 85. doi: 10.1002/ajmg.c.30087
55. Lopaschuk GD, et al. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2022; 90(1): 207 — 258.
56. Moore SJ, Haites NE, Broom I, White I, Coleman RJ, Pourfarzam M, et al. Acylcarnitine analysis in the investigation of myopathy. J Inherit Metab Dis. 1998; 21: 427 — 8.
57. Moorthie S, et al. Systematic review and meta-analysis to estimate the birth prevalence of five inherited metabolic diseases. J Inherit Metab Dis. 2022; 37(6): 889 — 898.
58. Oliveira SF, Pinho L, Rocha H, Nogueira C, Vilarinho L, Dinis MJ, Silva C. Rhabdomyolysis as a presenting manifestation of very long-chain acyl-coenzyme a dehydrogenase deficiency. Clin Pract. 2022 Aug 6; 3(2): e22.
59. Rashed MS, et al. Diagnosis of inborn errors of metabolism from blood spots by acylcarnitines and amino acids profiling using automated electrospray tandem mass spectrometry. Pediatr Res. 1995; 38(3): 324 — 331.
60. Saudubray JM, et al. Recognition and management of fatty acid oxidation defects: a series of 107 patients. J Inherit Metab Dis. 1999; 22(4): 488 — 502. doi: 10.1023/A:1005556207210.
61. Sharef Waadallah Sharef, Khalfan Al-Senaidi, and Surendra Nath Joshi. Successful Treatment of Cardiomyopathy due to Very Long-Chain Acyl-CoA Dehydrogenase Deficiency: First Case Report from Oman with Literature Review. Oman Medical Journal (2022) Vol. 28, No. 5: 354 — 356.
62. Solis JO, Singh RH. Management of fatty acid oxidation disorders: A survey of current treatment strategies. J Am Diet Assoc. 2002; 102: 1800 — 3.
63. Spiekerkoetter U, Bastin J., Gillingham M., Morris A // Current issues regarding treatment of mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis. 2022 Oct; 33(5): 555 — 61. doi: 10.1007/s10545-010-9188-1.
64. Spiekerkoetter U, et al. Lethal undiagnosed very long-chain acyl-CoA dehydrogenase deficiency with mild C14-Acylcarnitine abnormalities on newborn screening. JIMD Rep. 2022; 6: 113 — 115.
65. Spiekerkoetter U, et al. Management and outcome in 75 individuals with long-chain fatty acid oxidation defects: results from a workshop. J Inherit Metab Dis. 2009; 32(4): 488 — 497. doi: 10.1007/s10545-009-1125-9.
66. Spiekerkoetter U, Lindner M, Santer R, et al. Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop. J Inherit Metab Dis. 2009; 32: 498 — 505. doi: 10.1007/s10545-009-1126-8
67. Spiekerkoetter U, Mayatepek E. Update on mitochondrial fatty acid oxidation disorders. J Inherit Metab Dis. 2022; 33(5): 467 — 468. doi: 10.1007/s10545-010-9208-1.
68. Spiekerkoetter U. Mitochondrial fatty acid oxidation disorders: clinical presentation of long-chain fatty acid oxidation defects before and after newborn screening. J Inherit Metab Dis. 2022; 33(5): 527 — 532. doi: 10.1007/s10545-010-9090-x.
69. Tenopoulou M, Chen J, Bastin J, Bennett MJ, Ischiropoulos H, Doulias PT. Strategies for correcting very long chain acyl-CoA dehydrogenase deficiency. J Biol Chem. 2022 Apr 17; 290(16): 10486 — 94.
70. Topcu Y, et al. Importance of acylcarnitine profile analysis for disorders of lipid metabolism in adolescent patients with recurrent rhabdomyolysis: report of two cases. Ann Indian Acad Neurol. 2022; 17(4): 437 — 440.
71. Tucci S, Flogel U, Hermann S, Sturm M, Schafers M, Spiekerkoetter U. Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD (-/-)) mice. Biochim Biophys Acta. 2022 May; 1842(5): 677 — 85.
72. Wust RC, et al. Ketones and inborn errors of metabolism: old friends revisited. J Inherit Metab Dis. 2022; 40(1): 3 — 4.
73. Xiong D, He H, James J, et al. Cardiac-specific VLCAD deficiency induces dilated cardiomyopathy and cold intolerance. Am J Physiol Heart Circ Physiol. 2022 Feb; 306(3): H326 — 38.
74. Goetzman ES. Advances in the Understanding and Treatment of Mitochondrial Fatty Acid Oxidation Disorders. Curr Genet Med Rep. 2022; 5(3): 132 — 142. doi: 10.1007/s40142-017-0125-6
75. Pedersen CB, Kolvraa S, Kolvraa A, et al. The ACADS gene variation spectrum in 114 patients with short-chain acyl-CoA dehydrogenase (SCAD) deficiency is dominated by missense variations leading to protein misfolding at the cellular level. Hum Genet 2008; 124: 43 — 56. 10.1007/s00439-008-0521-9
76. Wolfe L, Jethva R, Oglesbee D, et al. Short-Chain Acyl-CoA Dehydrogenase Deficiency. 2022 Sep 22 [Updated 2022 Aug 9]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993 — 2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK63582/https://www.ncbi.nlm.nih.gov/books/NBK63582/
77. Martins E, Cardoso ML, Rodrigues E, et al. Short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: the clinical relevance of an early diagnosis and report of four new cases. J Inherit Metab Dis 2022; 34: 835 — 42. 10.1007/s10545-011-9287-7
78. Kapoor RR, James C, Flanagan SE, et al. 3-Hydroxyacyl-coenzyme A dehydrogenase deficiency and hyperinsulinemic hypoglycemia: characterization of a novel mutation and severe dietary protein sensitivity. J Clin Endocrinol Metab 2009; 94: 2221 — 5. 10.1210/jc.2009-0423
79. Clayton PT, Eaton S, Aynsley-Green A, et al. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. J Clin Invest 2001; 108: 457 — 65. 10.1172/JCI200111294
80. Tyni T, Ekholm E, Pihko H. Pregnancy complications are frequent in long-chain 3-hydroxyacyl-coenzyme a dehydrogenase deficiency. Am J Obstet Gynecol. 1998; 178(3): 603 — 608
81. «Nutrition Management of Inherited Metabolic Diseases» Laurie E. Bernstein, Fran Rohr, Joanna R. Helm 2022, Springer.com DOI 10.1007/978-3-319-14621-8 XIV
82. Magoulas, P.L., El-Hattab, A.W. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J Rare Dis 7, 68 (2022). https://doi.org/10.1186/1750-1172-7-68
83. Stanley CA, Bennett MJ, Longo N: Plasma membrane carnitine transporter defect. Online metabolic and molecular bases of inherited disease. Edited by Valle D, Beaudet AL, Vogelstein B, Kinzler KW, et al. 2006. http://www.ommbid.com/. Published January 2006. Updated March 28, 2022
84. Rubio-Gozalbo ME, Bakker JA, Waterham HR, Wanders RJ. Carnitine-acylcarnitine translocase deficiency, clinical, biochemical and genetic aspects. Mol Aspects Med. 2004; 25: 521 — 532. doi: 10.1016/j.mam.2004.06.007
85. Brivet M (2004) Carnitine-acylcarnitine translocase deficiency. Orphanet Encyclopedia 1 — 5
86. Lund AM, Skovby F, Vestergaard H, Christensen M, Christensen E. Clinical and biochemical monitoring of patients with fatty acid oxidation disorders. J Inherit Metab Dis. 2022; 33: 495 — 500. doi: 10.1007/s10545-009-9000-2
87. Vitoria I, Martin-Hernindez E, Pena-Quintana L, et al. Carnitine-acylcarnitine translocase deficiency: experience with four cases in Spain and review of the literature. JIMD Rep. 2022; 20: 11 — 20. doi: 10.1007/8904_2022_382
88. Iacobazzi V, Pasquali M, Singh R, et al. Response to therapy in carnitine/acylcarnitine translocase (CACT) deficiency due to a novel missense mutation. Am J Med Genet A. 2004; 126A: 150 — 155. doi: 10.1002/ajmg.a.20573
89. Spiekerkoetter U, Duran M. Mitochondrial fatty acid oxidation defects. In: Blau N, Duran M, Gibson KM, Dionisi-Visi C, editors. Physician’s guide to the diagnosis, treatment, and follow-up of inherited metabolic diseases. Berlin/Heidelberg: Springer-Verlag; 2022. pp. 247 — 264
90. Rubio-Gozalbo ME, Vos P, Forget PP, et al. Carnitine-acylcarnitine translocase deficiency: case report and review of the literature. Acta Paediatr. 2003; 92: 501 — 504. doi: 10.1111/j.1651-2227.2003.tb00586.x.
91. Bennett MJ, Santani AB. Carnitine Palmitoyltransferase 1A Deficiency. 2005 Jul 27 [Updated 2022 Mar 17]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993 — 2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1527/https://www.ncbi.nlm.nih.gov/books/NBK1527/
92. Angelini, C., Federico, A., Reichmann, H., Lombes, A., Chinnery, P. and Turnbull, D. (2006), Task force guidelines handbook: EFNS guidelines on diagnosis and management of fatty acid mitochondrial disorders. European Journal of Neurology, 13: 923 — 929. doi: 10.1111/j.1468-1331.2006.01482.x
93. Better OS, Stein GH. Early management of shock and prophylaxis of acute renal failure in traumatic rhabdomyolisis. The New England Journal of Medicine 1990; 322: 825 — 82
94. Merritt JL 2nd, Chang IJ. Medium-Chain Acyl-Coenzyme A Dehydrogenase Deficiency. 2000 Apr 20 [Updated 2022 Jun 27]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993 — 2020. Available from: https://www.ncbi.nlm.nih.gov/books/NBK1424/https://www.ncbi.nlm.nih.gov/books/NBK1424/
95. Lee PJ, Harrison EL, Jones MG, Jones S, Leonard JV, Chalmers RA. L-carnitine and exercise tolerance in medium-chain acyl-coenzyme A dehydrogenase (MCAD) deficiency: a pilot study. J Inherit Metab Dis. 2005; 28: 141 — 52.
96. Madsen KL, Preisler N, Orngreen MC, Andersen SP, Olesen JH, Lund AM, Vissing J. Patients with medium-chain acyl-coenzyme a dehydrogenase deficiency have impaired oxidation of fat during exercise but no effect of L-carnitine supplementation. J Clin Endocrinol Metab. 2022; 98: 1667 — 75.
97. Saud H. Aldubayan, Lance H. Rodan, Gerard T. Berry, Harvey L. Levy. Acute Illness Protocol for Fatty Acid Oxidation and Carnitine Disorders. Pediatric Emergency Care, 2022. V33, Number 4, p 300.
98. (Clinical paediatric dietetics / edited by Vanessa Shaw. — Fourth edition. Copyright 2022 John Wiley & Sons, Ltd, Print ISBN: 9780470659984 (Online ISBN: 9781118915349 (DOI: 10.1002/9781118915349)
99. [Lang TF. Adult presentations of medium-chain acyl-CoA dehydrogenase deficiency (MCADD). J Inherit Metab Dis. 2009; 32: 675 — 83].
100. [Uppala R, Dudiak B, Beck ME, Bharathi SS, Zhang Y, Stolz DB, Goetzman ES. Aspirin increases mitochondrial fatty acid oxidation. Biochem Biophys Res Commun. 2022; 482: 346 — 51].
101. Fraser H, Geppert J, Johnson R, et al. Evaluation of earlier versus later dietary management in long-chain 3-hydroxyacyl-CoA dehydrogenase or mitochondrial trifunctional protein deficiency: a systematic review. Orphanet J Rare Dis. 2022; 14(1): 258. Published 2022 Nov 15. doi: 10.1186/s13023-019-1226-y
102. Yamada, K., Taketani, T. Management and diagnosis of mitochondrial fatty acid oxidation disorders: focus on very-long-chain acyl-CoA dehydrogenase deficiency. J Hum Genet 64, 73 — 85 (2022). https://doi.org/10.1038/s10038-018-0527-7
103. Angle B, Burton BK. Risk of sudden death and acute life-threatening events in patients with glutaric acidemia type II. Mol Genet Metab 2008; 93: 36 — 9. 10.1016/j.ymgme.2007.09.015]
104. [Gharbawy A, Vockley J. Inborn Errors of Metabolism with Myopathy: Defects of Fatty Acid Oxidation and the Carnitine Shuttle System. Pediatr Clin North Am 2022; 65: 317 — 35. 10.1016/j.pcl.2022.11.006].
105. Merritt JL 2nd, Norris M, Kanungo S. Fatty acid oxidation disorders. Ann Transl Med. 2022; 6(24): 473. doi: 10.21037/atm.2022.10.57.
106. Bosch AM, Abeling NG, Ijlst L, et al. Brown-Vialetto-Van Laere and Fazio Londe syndrome is associated with a riboflavin transporter defect mimicking mild MADD: a new inborn error of metabolism with potential treatment. J Inherit Metab Dis. 2022; 34(1): 159 — 164. doi: 10.1007/s10545-010-9242-z.
107. Roe CR, Mochel F. Anaplerotic diet therapy in inherited metabolic disease: therapeutic potential. J Inherit Metab Dis. 2006; 29: 332 — 340. doi: 10.1007/s10545-006-0290-3.)
108. Derks TG, Reijngoud DJ, Waterham HR, Gerver WJ, Van Den Berg MP, Sauer PJ, Smit GP. The natural history of medium-chain acyl CoA dehydrogenase deficiency in the Netherlands: clinical presentation and outcome. J Pediatr. 2006; 148: 665 — 70
109. U. Spiekerkoetter & M. Lindner & R. Santer & M. Grotzke & M.R. Baumgartner & H. Boehles & A. Das & C. Haase & J.B. Hennermann & D. Karall & H. de Klerk & I. Knerr & H.G. Koch & B. Plecko & W. Roschinger & K.O. Schwab & D. Scheible & F.A. Wijburg & J. Zschocke & E. Mayatepek & U. Wendel. Treatment recommendations in long-chain fatty acid oxidationdefects: consensus from a workshop. May 2009 Journal of Inherited Metabolic Disease 32(4): 498 — 505. DOI: 10.1007/s10545-009-1126-8
110. J. Lawrence Merritt 2nd, Erin MacLeod, Agnieszka Jurecka & Bryan Hainline. Clinical manifestations and management of fatty acid oxidation disorders. Reviews in Endocrine and Metabolic Disorders volume 21, pages 479 — 493 (2020).
111. Prasun P. Multiple Acyl-CoA Dehydrogenase Deficiency. 2020 Jun 18. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews [Internet]. Seattle (WA): University of Washington, Seattle; 1993 — 2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK558236/
112. Gillingham et al. 2006; Spiekerkoetter 2007.
113. Boer den MEJ, Wanders RJA, Morris AAM, IJLst L, Long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency: clinical presentation and follow-up of 50 patients. Pediatrics. 2002; 109(1): 99 — 104
114. Boer den MEJ, Dionisi-Vici C, Mitochondrial trifunctional protein deficiency: a severe fatty acid oxidation disorder with cardiac and neurologic involvement. J Pediatr. 2003; 142(6): 684 — 689. doi: 10.1067/mpd.2003.231
115. Tyni T, Johnson M, Eaton S, Pourfarzam M, Andrews R, Turnbull DM. Mitochondrial fatty acid beta-oxidation in the retinal pigment epithelium. Pediatric Research. 2002; 52(4): 595 — 600. doi: 10.1203/00006450-200210000-00021
116. Oey NA, Ruiter JPN, Attie-Bitach T, IJLst L, Wanders RJA, Wijburg FA. Fatty acid oxidation in the human fetus: implications for fetal and adult disease. J Inherit Metab Dis. 2006; 29(1): 71 — 75. doi: 10.1007/s10545-006-0199-x
117. Fletcher AL, Pennesi ME, harding CO, Weleber RG, Gillingham MB. Observations regarding retinopathy in mitochondrial trifunctional protein deficiencies. Molecular Genetics and Metabolism. 2022. doi: 10.1016/j.ymgme.2022.02.015
118. Makhseed N., Vallance H.D., Potter M. et all. Carnitine transporter defect due to a novel mutation in the SLC22A5 gene presenting with peripheral neuropathy//J.Inherit.Metab.Dis.27 (2004): 778 — 780; https://doi.org/10.1023/B:BOLI.0000045837.23328.f4
119. Brivet M., et all. Defects in activation and transport of fatty acids. 1999. https://doi.org/10.1023/A:1005552106301
120. Vianey-Saban C., Stremler N., Paut O. et al. (1995). Infantile form of carnitine palmitoyltransferase II deficiency in a girl with rapid fatal onset. J Inherit Metab Dis 18: 362 — 363.
121. Taroni F., Verderio E., Fiorucci S., et al. (1992). Molecular characterization of inherited carnitine palmitoyltransferase II deficiency. Proc Natl Acad Sci USA 89: 8429 — 8433.
122. North K.N., Hoppel C.L. et all Lethal neonatal deficiency of carnitine palmitoiltransferase II associated with dysgenesis of the brain and kidneys. J. Pediatr 1995; 127: 414
123. Mathur A., Sims H.F., Gopalakrishnan D. et al. Molecular heterogeneity in very long chain acyl CoA dehydrogenase deficiency causing pediatric cardiomyopathy and sudden death. Circulation 1999; 99: 1337 — 1343.
124. Dipti S, Childs AM, Livingston JH, Aggarwal AK, Miller M, Williams C, et al. Brown-Vialetto-Van Laere syndrome; variability in age at onset and disease progression highlighting the phenotypic overlap with Fazio-Londe disease. Brain Dev. 2005; 27: 443 — 446.
125. O’Callaghan B, Bosch AM, Houlden H. An update on the genetics, clinical presentation, and pathomechanisms of human riboflavin transporter deficiency. J Inherit Metab Dis. 2022; 42: 598 — 607.
126. Olsen R.K., Konarikova E., Giancaspero T.A. et al. Riboflavin-responsive and -non-responsive mutations in FAD synthase cause multiple acyl-CoA dehydrogenase and combined respiratory-chain deficiency. Am J Hum Genet. 2022; 98: 1130 — 1145.
127. Schiff M., Veauville-Merllie A., Su C.H. et al. SLC25A32 mutations and riboflavin-responsive exercise intolerance. N Engl J Med. 2022; 374: 795 — 797
128. Hellebrekers D.M., Sallevelt S.C., Theunissen T.E. et al. Novel SLC25A32 mutation in a patient with a severe neuromuscular phenotype. Eur J Hum Genet. 2022; 25: 886 — 888.
129. Sklirou E., Alodaib A.N., Dobrowolski S.F. et al. Physiological Perspectives on the Use of Triheptanoin as Anaplerotic Therapy for Long Chain Fatty Acid Oxidation Disorders//Front. Genet., 15 January 2021 / https://doi.org/10.3389/fgene.2020.598760
130. Helene Ogier de Baulny, Andrea Superti-Furga. Disorders of mitochondrial fatty acid oxidation and ketone body metabolism. In: Blau N., Hoffmann G.F., Leonard J.V., Clarke J.T.R. Physician’s Guide to the Treatment and Follow-Up of Metabolic Diseases. Springer Science & Business Media, pp 147 — 160 (http://eknygos.lsmuni.lt/springer/365/147-160.pdf)
131. А.В. Дегтярева, И.В. Никитина, И.В. Орловская, Е.Ю. Захарова, Г.В. Байдакова, О.В. Ионов, Д.Ю. Амирханова, А.В. Левадная. Дефицит ацил-коэнзим ФА дегидрогеназы жирных кислот с очень длинной углеродной цепью
132. Amaral, Alexandre U., Cecatto, Cristiane, Silva, Janaina C. da, Wajner, Alessandro, & Wajner, Moacir. (2022). Mechanistic Bases of Neurotoxicity Provoked by Fatty Acids Accumulating in MCAD and LCHAD Deficiencies. Journal of Inborn Errors of Metabolism and Screening, 5, e160052. Epub May 16, 2022
133. Smith E, Fernandez C, Melander O, Ottosson F. Altered Acylcarnitine Metabolism Is Associated With an Increased Risk of Atrial Fibrillation. J Am Heart Assoc. 2020 Nov 3; 9(21): e016737, https://emedicine.medscape.com/article/2087425-overview#a4
134. Gillingham MB, Purnell JQ, Jordan J, Stadler D, Haqq AM, Harding CO. Effects of higher dietary protein intake on energy balance and metabolic control in children with long-chain 3-hydroxy acyl-CoA dehydrogenase (LCHAD) or trifunctional protein (TFP) deficiency. Mol Genet Metab. 2007 Jan; 90(1): 64 — 9. doi: 10.1016/j.ymgme.2006.08.002.
135. DiDonato S, Gellera C, Peluchetti D, Uziel G, Antonelli A, Lus G, Rimoldi M Normalization of short-chain acylcoenzyme A dehydrogenase after riboflavin treatment in a girl with multiple acylcoenzyme A dehydrogenase-deficient myopathy
136. Kmoch S., Zeman J, Hrebicek M, Ryba L, Kristensen MJ, Gregersen N.J Inherit Metab Dis Riboflavin-responsive epilepsy in a patient with SER209 variant form of short-chain acyl-CoA dehydrogenase. 1995; 18(2): 227 — 9. doi: 10.1007/BF00711774.
137. Harding C.O., Gillingham M.B., van Calcar S.C., Wolff J.A., Verhoeve J.N., Mills M.D. Docosahexaenoic acid and retinal function in children with long-chain 3-hydroxyacyl-CoA dehydrogenase deficiency. 1999. May; 22(3): 276 — 80. doi: 10.1023/a:1005502626406.
138. Sun XL, Yang Z, Wang JL, Sun MN, Wu SY, Wang XY. [Correlation between severe preeclampsia and abnormal expression of long-chain fatty acid oxidative enzyme]. Zhonghua Yi Xue Za Zhi. 2022 Aug 9; 91(29): 2026 — 9. Chinese. PMID: 22093928.
139. https://www.awmf.org/uploads/tx_szleitlinien/027-006l_S3_Diagnostik-Therapie-Harnstoffzyklusstoerungen_2022-06.pdf
140. https://ojrd.biomedcentral.com/articles/10.1186/s13023-014-0130-8/tables/7
141. Дегтярева А.В., Соколова Е.В., Захарова Е.Ю., Исаева М.Х., Высоких М.Ю., Иванец Т.Ю., Дегтярев Д.Н. Гипераммониемия в практике неонатолога. Рос вестн перинатол и педиатр 2020; 65: (6): 98 — 107. DOI: 10.21508/1027-4065-2020-65-6-98-107
142. Knottnerus, S.J.G., Bleeker, J.C., Wust, R.C.I. et al. Disorders of mitochondrial long-chain fatty acid oxidation and the carnitine shuttle. Rev Endocr Metab Disord 19, 93 — 106 (2022). https://doi.org/10.1007/s11154-018-9448-1
143. Zhu, M., Zhu, X., Qi, X., Weijiang, D., Yu, Y., Wan, H., & Hong, D. (2022). Riboflavin-responsive multiple Acyl-CoA dehydrogenation deficiency in 13 cases and a literature review in mainland Chinese patients. Journal of Human Genetics, 59(5), 256 — 261. doi: 10.1038/jhg.2022.10
144. Wanders RJ, Visser G, Ferdinandusse S, Vaz FM, Houtkooper RH. Mitochondrial Fatty Acid Oxidation Disorders: Laboratory Diagnosis, Pathogenesis, and the Complicated Route to Treatment. J Lipid Atheroscler. 2020 Sep; 9(3): 313 — 333. https://doi.org/10.12997/jla.2020.9.3.313
145. Kompare M. & Rizzo W. B. Mitochondrial Fatty-Acid Oxidation Disorders. Seminars in Pediatric Neurology. 2008. 15(3), 140 — 149. doi: 10.1016/j.spen.2008.05.008
146. El-Hattab, Ayman W; Li, Fang-Yuan; Shen, Joseph; Powell, Berkley R; Bawle, Erawati V; Adams, Darius J; Wahl, Erica; Kobori, Joyce A; Graham, Brett; Scaglia, Fernando; Wong, Lee-Jun (2022). Maternal systemic primary carnitine deficiency uncovered by newborn screening: Clinical, biochemical, and molecular aspects. Genetics in Medicine, 12(1), 19 — 24. doi: 10.1097/GIM.0b013e3181c5e6f7
147. Lisa A. Schimmenti; Eric A. Crombez; Bernd C. Schwahn; Bryce A. Heese; Timothy C. Wood; Richard J. Schroer; Kristi Bentler; Stephen Cederbaum; Kiki Sarafoglou; Mark McCann; Piero Rinaldo; Dietrich Matern; Cristina Amat di San Filippo; Marzia Pasquali; Susan A. Berry; Nicola Longo (2007). Expanded newborn screening identifies maternal primary carnitine deficiency. 90(4), 0 — 445. doi: 10.1016/j.ymgme.2006.10.003
148. S.E. Olpin, J. Allen, J.R. Bonham et al. Features of carnitine palmitoyltransferase type I deciency J. Inherit. Metab. Dis. 24 (2001) 35 — 42
149. Nyhan, W.L., Hoffmann, G.F., Al-Aqeel, A.I., & Barshop, B.A. (2022). Atlas of Inherited Metabolic Diseases (4th ed.). CRC Press. https://doi.org/10.1201/9781315114033
150. F Feillet, G Steinmann, C Vianey-Saban, C de Chillou, N Sadoul, E Lefebvre, M Vidailhet, PE Bollaert Adult presentation of MCAD deficiency revealed by coma and severe arrythmias Intensive Care Med (2003) 29: 1594 — 1597 DOI 10.1007/s00134-003-1871-3
151. https://www.newenglandconsortium.org/mcadd
152. Savy N, Brossier D, Brunel-Guitton C, Ducharme-Crevier L, Du Pont-Thibodeau G, Jouvet P. Acute pediatric hyperammonemia: current diagnosis and management strategies. Hepat Med. 2022 Sep 12; 10: 105 — 115. doi: 10.2147/HMER.S140711. PMID: 30254497; PMCID: PMC6140721.
153. https://www.uptodate.com/contents/insulin-regular-pediatric-drug-information?search=insulin&topicRef=85908&source=see_link
154. Matoori S, Leroux JC. Recent advances in the treatment of hyperammonemia. Adv Drug Deliv Rev. 2022 Aug 1; 90: 55 — 68.
155. Magoulas PL, El-Hattab AW. Systemic primary carnitine deficiency: an overview of clinical manifestations, diagnosis, and management. Orphanet J Rare Dis. 2022; 7: 68.
156. El-Hattab AW. Systemic Primary Carnitine Deficiency. In: GeneReviews. University of Washington, Seattle, Seattle (WA); 1993. PMID: 22420015.
157. Gillingham MB, Weleber RG, Neuringer M, et al. Effect of optimal dietary therapy upon visual function in children with long-chain 3-hydroxyacyl CoA dehydrogenase and trifunctional protein deficiency. Mol Genet Metab. 2005 Sep — Oct; 86(1-2): 124 — 33.
158. Gataullina S, Delonlay P, Lemaire E, et al. Seizures and epilepsy in hypoglycaemia caused by inborn errors of metabolism. Dev Med Child Neurol. 2022 Feb; 57(2): 194 — 9.
Приложение А1
СОСТАВ РАБОЧЕЙ ГРУППЫ ПО РАЗРАБОТКЕ И ПЕРЕСМОТРУ
КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЙ
1. Багаева Мадлена Энверовна — к.м.н., старший научный сотрудник отделения педиатрической гастроэнтерологии, гепатологии и диетотерапии ФГБУН «ФИЦ питания и биотехнологии», ассистент кафедры ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России.
2. Байдакова Галина Викторовна — к.б.н., ведущий научный сотрудник лаборатории наследственных болезней обмена ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова», член Российского общества медицинских генетиков.
3. Баранов Александр Александрович — акад. РАН, профессор, д.м.н.; почетный президент Союза педиатров России, советник руководителя НИИ педиатрии и охраны здоровья детей ЦКБ РАН, профессор кафедры педиатрии и детской ревматологии ФГАОУ «Первый МГМУ им. И.М. Сеченова» Минздрава России (Сеченовский Университет), главный внештатный специалист педиатр Минздрава России.
4. Бушуева Татьяна Владимировна — д.м.н., ФГАУ «Научный медицинский исследовательский центр здоровья детей» МЗ РФ, член Союза педиатров России.
5. Вашакмадзе Нато Джумберовна — д.м.н., руководитель отдела орфанных болезней и профилактики инвалидизирующих заболеваний НИИ педиатрии и охраны здоровья детей ЦКБ РАН профессор кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России.
6. Вишнева Елена Александровна — д.м.н., заместитель руководителя НИИ педиатрии и охраны здоровья детей ЦКБ РАН Минобрнауки по научной работе, доцент кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России, член Союза педиатров России.
7. Дегтярева Анна Владимировна — д.м.н., профессор, заведующая отделом педиатрии, ФГБУ «НМИЦ акушерства, гинекологии и перинатологии им. Ак. В.И. Кулакова», член Российского общества неонатологов.
8. Докшукина Алина Алексеевна — врач-неонатолог, младший научный сотрудник отдела педиатрии ФГБУ «НМИЦ акушерства, гинекологии и перинатологии им. Ак. В.И. Кулакова» член Российского общества неонатологов.
9. Зарубина Вера Владимировна — врач-генетик Морозовской ДГКБ ДЗМ.
10. Журкова Наталия Вячеславовна — к.м.н., ведущий научный сотрудник НИИ педиатрии и охраны здоровья детей ЦКБ РАН, член Союза педиатров России, член Ассоциации медицинских генетиков (АМГ).
11. Захарова Екатерина Юрьевна — д.м.н., заведующая лабораторией наследственных болезней обмена ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова», член Российского общества медицинских генетиков, член европейского общества по изучению наследственных болезней обмена веществ (SSIEM).
12. Кузенкова Людмила Михайловна — д.м.н., заведующая отделением психоневрологии и психосоматической патологии ФГАУ «Научный медицинский исследовательский центр здоровья детей» МЗ РФ, член Союза педиатров России.
13. Куцев Сергей Иванович — чл.-корр РАН, д.м.н., директор ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова», Президент Ассоциации медицинских генетиков (АМГ).
14. Лаврова Алла Евгеньевна — д.м.н., главный научный сотрудник, врач высшей квалификационной категории по специальности «Педиатрия», заведующая 2-м педиатрическим отделением, ФГБОУ ВО «Приволжский исследовательский медицинский университет» МЗ РФ.
15. Михайлова Светлана Витальевна — д.м.н., заведующая отделением ФГБУ «Российская Детская Клиническая Больница» МЗ РФ.
16. Намазова-Баранова Лейла Сеймуровна — акад. РАН, профессор, д.м.н., президент Союза педиатров России; паст-президент EPA/UNEPSA; руководитель НИИ педиатрии и охраны здоровья детей ЦКБ РАН, заведующая кафедрой факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России, главный внештатный детский специалист по профилактической медицине Минздрава России.
17. Назаренко Людмила Павловна — профессор, д.м.н., заместитель директора по научной и лечебной работе, руководитель лаборатории наследственной патологии НИИ медицинской генетики, Томского НИМЦ РАН, член Ассоциации медицинских генетиков (АМГ).
18. Николаева Екатерина Александровна — д.м.н., главный научный сотрудник отделения наследственных и врожденных заболеваний с нарушением психики Научно-исследовательского клинического института педиатрии имени акад. Ю.Е. Вельтищева ГБОУ ВПО РНИМУ им. Н.И. Пирогова Минздрава России отдела клинической генетики Научно-исследовательского клинического института педиатрии имени акад. Ю.Е. Вельтищева ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России.
19. Первунина Татьяна Михайловна — д.м.н., врач-педиатр, врач-детский кардиолог, доцент кафедры педиатрии медицинского факультета СПБГУ, директор института педиатрии и перинатологии ФГБУ «НМИЦ им. В.А. Алмазова».
20. Печатникова Наталья Леонидовна — руководитель Городского Центра орфанных и других редких заболеваний у детей и подростков ГБУЗ «Морозовская ДГКБ ДЗМ».
21. Репина Светлана Афанасьевна — к.м.н., врач-генетик ФГБНУ «Медико-генетический научный центр им. академика Н.П. Бочкова», член Российского общества медицинских генетиков, член Ассоциации медицинских генетиков.
22. Смирнова Ольга Яковлевна — врач-генетик, старший научный сотрудник отдела стандартизации и изучения основ доказательной медицины НИИ педиатрии и охраны здоровья детей ЦКБ РАН.
23. Сеитова Гульнара Наримановна — к.м.н., главный врач Медико-генетического центра (Генетической клиники) НИИ медицинской генетики, Томского НИМЦ РАН.
24. Строкова Татьяна Викторовна — д.м.н., профессор РАН, заведующая отделением педиатрической гастроэнтерологии, гепатологии и диетотерапии ФГБУН «ФИЦ питания и биотехнологии», заведующая кафедрой «Гастроэнтерологии и диетологии» ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России.
25. Селимзянова Лилия Робертовна — к.м.н., ведущий научный сотрудник НИИ педиатрии и охраны здоровья детей ЦКБ РАН Министерства науки и высшего образования Российской Федерации, доцент кафедры педиатрии и детской ревматологии ФГАОУ «Первый МГМУ им. И.М. Сеченова» Минздрава России (Сеченовский Университет), доцент кафедры факультетской педиатрии педиатрического факультета ФГБОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России, член Союза педиатров России.
26. Таран Наталия Николаевна — к.м.н., старший научный сотрудник отделения педиатрической гастроэнтерологии, гепатологии и диетотерапии ФГБУН «ФИЦ питания и биотехнологии», ассистент кафедры ФГАОУ ВО РНИМУ им. Н.И. Пирогова Минздрава России.
27. Фисенко Андрей Петрович — д.м.н., профессор, Заслуженный врач Российской Федерации, директор ФГАУ «Национальный медицинский исследовательский центр здоровья детей» Минздрава России.
Представители общественных пациентских организаций:
Погосян Нелли Сергеевна — представитель Всероссийского общества орфанных заболеваний
Авторы подтверждают отсутствие финансовой поддержки/конфликта интересов, который необходимо обнародовать.
Приложение А2
МЕТОДОЛОГИЯ РАЗРАБОТКИ КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЙ
Настоящие рекомендации предназначены для применения медицинскими организациями и учреждениями федеральных, территориальных и муниципальных органов управления здравоохранением, систем обязательного и добровольного медицинского страхования, другими медицинскими организациями различных организационно-правовых форм деятельности, направленной на оказание медицинской помощи.
Настоящие рекомендации устанавливают виды, объем и индикаторы качества медицинской помощи пациентам при нарушениях окисления жирных кислот и были рассмотрены 1 — 2 июня 2022 года в рамках Научно-практического конгресса «Орфанные болезни» в Москве.
Клинические рекомендации созданы на основании систематического обзора литературы 1992 — 2022 гг. Medline (Pubmed version), Embase (Dialog version) и Cochrane Library databases, с использованием созданных протоколов современных международных клинических рекомендаций по диагностике, лечению и ведению пациентов с метаболическими болезнями.
Нарушения окисления жирных кислот относятся к редким наследственным заболеваниям, что исключает возможность проведения больших когортных и рандомизированных контролированных исследований и для создания протоколов диагностики и терапии используются лишь тематические исследования экспертов, опубликованные в последние два десятилетия.
Оценка качества доказательств и силы рекомендаций применения медицинских технологий проводилась в соответствии с унифицированной шкалой, приведенной в таблицах 1 — 3.
Целевая аудитория данных клинических рекомендаций:
1. Врачи общей практики (семейные врачи);
2. Врачи-педиатры;
3. Врачи-терапевты;
4. Врачи-генетики;
5. Врачи-лабораторные генетики;
6. Врачи-кардиологи;
7. Врачи-детские кардиологи;
8. Врачи-неврологи;
9. Врачи функциональной диагностики;
10. Медицинские психологи;
11. Студенты медицинских ВУЗов;
12. Обучающиеся в ординатуре и интернатуре.
Таблица 1. Шкала оценки уровней достоверности доказательств (УДД) для методов диагностики (диагностических вмешательств)
Таблица 2. Шкала оценки уровней достоверности доказательств (УДД) для методов профилактики, лечения и реабилитации (профилактических, лечебных, реабилитационных вмешательств)
Таблица 3. Шкала оценки уровней убедительности рекомендаций (УУР) для методов профилактики, диагностики, лечения и реабилитации (профилактических, диагностических, лечебных, реабилитационных вмешательств)
Порядок обновления клинических рекомендаций.
Механизм обновления клинических рекомендаций предусматривает их систематическую актуализацию — не реже чем один раз в три года, а также при появлении новых данных с позиции доказательной медицины по вопросам диагностики, лечения, профилактики и реабилитации конкретных заболеваний, наличии обоснованных дополнений/замечаний к ранее утвержденным КР, но не чаще 1 раза в 6 месяцев.
Приложение А3
СПРАВОЧНЫЕ МАТЕРИАЛЫ, ВКЛЮЧАЯ СООТВЕТСТВИЕ ПОКАЗАНИЙ
К ПРИМЕНЕНИЮ И ПРОТИВОПОКАЗАНИЙ, СПОСОБОВ ПРИМЕНЕНИЯ И ДОЗ
ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ, ИНСТРУКЦИИ ПО ПРИМЕНЕНИЮ
ЛЕКАРСТВЕННОГО ПРЕПАРАТА
1. Приказ Министерства здравоохранения РФ «Об утверждении Порядка оказания медицинской помощи больным с врожденными и (или) наследственными заболеваниями» от 15 ноября 2022 г. N 917н). Федеральный закон «Об основах охраны здоровья граждан в Российской Федерации» (N 323-ФЗ от 21.11.2022).
2. Постановление Правительства РФ от 30 июля 1994 г. N 890 «О государственной поддержке развития медицинской промышленности и улучшении обеспечения населения и учреждений здравоохранения лекарственными средствами и изделиями медицинского назначения» (с изменениями и дополнениями)
3. Приказ Минздравсоцразвития РФ от 22.03.2006 N 185 «О массовом обследовании новорожденных детей на наследственные заболевания» (вместе с Положением об организации проведения массового обследования новорожденных детей на наследственные заболевания, Рекомендациями по забору образцов крови при проведении массового обследования новорожденных детей на наследственные заболевания)
4. Приказ Министерства здравоохранения Российской Федерации от 15 декабря 2022 г. N 834н «Об утверждении унифицированных форм медицинской документации, используемых в медицинских организациях, оказывающих медицинскую помощь в амбулаторных условиях, и порядков по их заполнению».
5. Приказ Министерства здравоохранения Российской Федерации от 20 декабря 2022 г. N 1177н «Об утверждении порядка дачи информированного добровольного согласия на медицинское вмешательство и отказа от медицинского вмешательства в отношении определенных видов медицинских вмешательств, форм информированного добровольного согласия на медицинское вмешательство и форм отказа от медицинского вмешательства».
6. Приказ Министерства здравоохранения Российской Федерации от 14 января 2022 г. N 4н «Об утверждении порядка назначения лекарственных препаратов, форм рецептурных бланков на лекарственные препараты, порядка оформления указанных бланков, их учета и хранения»
7. Приказ Министерства здравоохранения Российской Федерации от 16 мая 2022 г. N 302н «Об утверждении Порядка прохождения несовершеннолетними диспансерного наблюдения, в том числе в период обучения и воспитания в образовательных организациях»
8. Распоряжение Правительства РФ от 31 декабря 2022 г. N 3053-р «Об утверждении перечней медицинских изделий, имплантируемых в организм человека при оказании медицинской помощи в рамках программы государственных гарантий бесплатного оказания гражданам медицинской помощи и отпускаемых по рецептам на медицинские изделия при предоставлении набора социальных услуг»
Информация о лекарственных средствах: https://grls.rosminzdrav.ru/
Основные нормативно-правовые акты, регулирующие оказание паллиативной медицинской помощи
1. Федеральный закон «О внесении изменений в Федеральный закон «Об основах охраны здоровья граждан в Российской Федерации» по вопросам оказания паллиативной медицинской помощи» от 06.03.2022 N 18-ФЗ.
2. Приказ Минздрава России N 345н, Минтруда России от 31.05.2022 N 372н «Об утверждении положения об организации оказания паллиативной медицинской помощи, включая порядок взаимодействия медицинских организаций, организаций социального обслуживания и общественных объединений, иных некоммерческих организаций, осуществляющих свою деятельность в сфере охраны здоровья».
3. Приказ Минздрава России N 348н от 31 мая 2022 года «Об утверждении перечня медицинских изделий, предназначенных для поддержания органов и систем организма человека, предоставляемых для использования на дому».
4. Приказ Минздрава России N 505н от 10 июля 2022 года «Об утверждении Порядка передачи от медицинской организации пациенту (его законному представителю) медицинских изделий, предназначенных для поддержания функций органов и систем организма человека, для использования на дому при оказании паллиативной медицинской помощи».
5. Приказ МЗ РФ N 831 от 3 октября 2022 года «Об утверждении ведомственной целевой программы «Развитие системы оказания паллиативной медицинской помощи».
Прочие информационные ресурсы и нормативно-правовые документы, с учетом которых разработаны клинические рекомендации:
6. Международная классификация болезней, травм и состояний, влияющих на здоровье (МКБ-10);
7. Приказ МЗ РФ от 20 декабря 2022 г. N 1183н «Об утверждении номенклатуры должностей медицинских работников и фармацевтических работников».
8. Приказ МЗ РФ от 23 июля 2022 г. N 541н. Единый квалификационный справочник должностей руководителей, специалистов и служащих, раздел Квалификационные характеристики должностей работников в сфере здравоохранения.
9. Федеральный закон от 25.12.2022 N 489-ФЗ «О внесении изменений в статью 40 Федерального закона «Об обязательном медицинском страховании в Российской Федерации» и Федеральный закон «Об основах охраны здоровья граждан в Российской Федерации» по вопросам клинических рекомендаций».
10. Приказ Минздрава России N 103н от 28.02.2022 «Об утверждении порядка и сроков разработки клинических рекомендаций, их пересмотра, типовой формы клинических рекомендаций и требований к их структуре, составу и научной обоснованности, включаемой в клинические рекомендации информации».
11. Приказ Минздрава России от 13.10.2022 N 804н «Об утверждении номенклатуры медицинских услуг».
12. Приказ Министерства труда и социальной защиты РФ от 27 августа 2022 г. N 585н «О классификациях и критериях, используемых при осуществлении медико-социальной экспертизы граждан федеральными государственными учреждениями медико-социальной экспертизы».
13. Приказ Министерства здравоохранения и социального развития Российской Федерации «О порядке применения лекарственных средств у больных по жизненным показаниям» от 9 августа 2005 г. N 494.
14. Информационное письмо Минздрава России по возможности закупки лекарственного препарата по торговому наименованию (https://www.rosminzdrav.ru/news/2022/12/18/13043-minzdrav-podgotovil-informatsionnoe-pismo-po-vozmozhnosti-zakupki-lekarstvennogo-preparata-po-torgovomu-naimenovaniyu);
15. Перечень специализированных продуктов лечебного питания для детей-инвалидов на 2020 год, Распоряжение Правительства Российской Федерации от 11 декабря 2022 года N 2984-р.
Приложение Б
АЛГОРИТМЫ ДЕЙСТВИЙ ВРАЧА
Приложение В
ИНФОРМАЦИЯ ДЛЯ ПАЦИЕНТА
МЕТАБОЛИЗМ И НАРУШЕНИЯ ОБМЕНА ВЕЩЕСТВ
Термин «метаболизм» подразумевает под собой все процессы необходимые для функционирования и развития организма, в ходе которых образуется и расходуется энергия. Белки, жиры и углеводы — основные пищевые вещества, которые используются в качестве сырья для синтеза новых клеток или окисляются, доставляя организму энергию.
Процессы метаболизма происходят в человеческом организме ежесекундно. Даже в условиях полного покоя человек использует некоторое количество энергии. Она непрерывно тратится на физиологические процессы, которые не останавливаются ни на минуту.
Часть энергии необходима для построения новых тканевых компонентов, другая расходуется в процессе функционирования клеток: при сокращении мышц, передаче нервных импульсов. Остальная энергия освобождается в виде тепла. Для производства энергии мы используем продукты питания, а в их отсутствие резервные запасы питательных веществ, присутствующие в нашем организме. За каждый этап (биохимическую реакцию) в производстве энергии отвечает конкретный белок, фермент. У каждого из них есть своя функция. Это целый конвейер химических реакций. Если один фермент не работает, то вся цепочка останавливается, и организм не получает конечный продукт, что приводит к нарушению метаболизма, развитию заболеваний из группы наследственных нарушений обмена веществ (НБО).
МЕТАБОЛИЗМ ЖИРОВ
Жиры (триглицериды) это соединения карбоновых кислот и трехатомного спирта глицерина. Существуют различные типы жиров. В зависимости от типа жирных кислот, которые преимущественно образуют жиры, и от степени ненасыщенности (количества двойных или тройных связей) жирных кислот, различают: насыщенные жиры (твердые) — жиры животных и птиц, и ненасыщенные жиры (жидкие) — они встречаются в большом количестве в растительных маслах, орехах или морской рыбе.
Насыщенные жиры становятся ключевым источником энергии для организма в тех ситуациях, когда он подвергается сильным физическим нагрузкам. Ненасыщенные жирные кислоты участвуют в регенерации клеток и в иммунной защите. Одной из важнейших функций жира является энергетическая. Жир обеспечивает вдвое больше калорий (энергии) чем углеводы.
Рис. 1. Классификация жирных кислот
-ОКИСЛЕНИЕ ЖИРНЫХ КИСЛОТ
Митохондриальное окисление жирных кислот — один из главных источников энергии, обеспечивающий в период голодания до 80% от общей потребности. Когда уровень глюкозы снижается, а запасов гликогена становится недостаточно, чтобы покрыть все энергетические потребности (при голодании и длительной физической нагрузке) активируется липолиз. Происходит образование длинноцепочечных жирных кислот (C16 — C20) и глицерина из запасенных в жировой ткани триацилглицеринов. Регулирует липолиз гормончувствительная липаза, которая активируется гормонами, секретируемыми при голодании и стрессе: глюкагоном и кортизолом.
В печени, в митохондриях, идет процесс образования кетоновых тел — митохондриальное в-окисление жирных кислот. Кетоновые тела являются важнейшим источником энергии для головного мозга.
Для в-окисления жирные кислоты необходимо доставить в митохондрии. Внутренняя митохондриальная мембрана непроницаема для длинноцепочечных жирных кислот, и они проникают внутрь с помощью карнитиновой челночной системы. Карнитин-зависимые ферменты — карнитинтранслоказа, карнитинпальмитоилтрансферазы I и II — обеспечивают перенос длинноцепочечных жирных кислот в виде сложных эфиров (ацилкарнитинов) из цитоплазмы через наружную и внутреннюю митохондриальную мембрану в матрикс митохондрий. В процессе окисления жирных кислот, с каждым шагом их цепи укорачиваются, за каждый этап на этом пути несет ответственность определенный фермент.
Рис. 2. Схема в-окисления жирных кислот.
В цитоплазме длинноцепочечные жирные кислоты связываются с карнитином ферментом карнитин пальмитоилтрансферазой I (CPTI), образуя ацилкарнитины.
Ацилкарнитин транспортируется ферментом-транспортером карнитин-транслоказой (CACT) в митохондрии.
Ацилкарнитин снова расщепляется ферментом карнитин пальмитоилтрансферазой II (CPTII), высвобождая карнитин, который может вернуться в клетку и повторно использовать. Жирные кислоты с длинной цепью укорачивается до среднецепочечных жирных кислот ферментами VLCAD, LCHAD и TFTPFP. Укорочение жирных кислот называется -окислением
НАРУШЕНИЯ В-ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
При недостаточности ферментов, которые помогают жирным кислотам проникнуть в митохондрии (то место где идет синтез нужных нашему организму кетоновых тел), и дефиците ферментов, которые расщепляют жирные кислоты происходит блокирование (или резкое снижение) митохондриального в-окисления, что заставляет наш организм искать альтернативные пути утилизации жирных кислот, и это приводит к накоплению токсических веществ, которые оказывают неблагоприятное воздействие на ткани головного мозга, сердца, печени и др.
В норме активация митохондриального в-окисление жирных кислот, это физиологическая реакция организма на голодание или избыточное поступление жирной пищи, инфекционный процесс и повышенную физическую (с высокой мышечной активностью) или эмоциональную нагрузку. Но при патологии (нарушении процесса окисления жирных кислот). Это ведет к метаболическому ацидозу, гипераммониемии, усугублению гипогликемии, поражению внутренних органов.
Рис. 3. Схема в-окисления жирных кислот в норме и при патологии
КЛАССИФИКАЦИЯ НАРУШЕНИЙ В-ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ И ТРАНСПОРТА КАРНИТИНА
Известно более 15 различных форм нарушений митохондриального в-окисления жирных кислот. Все они связаны с нарушением белков или ферментов, которые участвуют в метаболизме жирных кислот. Поскольку названия ферментов очень длинные и сложные, принято использовать сокращенную аббревиатуру, английскую или русскую (в русских статьях и книгах).
1. Нарушение транспорта жирных кислот
— Первичный дефицит карнитина (ген SLC22A5)
Клинические проявления: кардиомиопатия, аритмия, сердечная недостаточность, мышечная слабость, поражение печени.
— Карнитин пальмитоилтрансферазы 1 дефицит (ген CPTI)
Клинические проявления: тяжелое поражение печени, нарушение ее функции, холестаз.
— Карнитин транслоказы дефицит (ген SLC25A20)
Клинические проявления: тяжелая кардимиопатия, аритмии, поражение печени.
— Карнитин пальмитоилтрансферазы 2 дефицит (ген CPTII)
Клинические проявления: при ранней манифестации (неонатальная форма) кардиомиопатия, поражение печени.
Поздняя манифестация, у взрослых, более мягкая форма, проявляется эпизодами мышечной слабости, трудностями при выполнении физической нагрузки.
2. Нарушение в-окисления жирных кислот
— Короткоцепочечной ацил-КоA дегидрогеназы дефицит (SCAD) (ген ACADS): обычно протекает бессимптомно.
— Среднецепочечной ацил-КоA дегидрогеназы дефицит MCAD (ген ACADM)
Клинические проявления: может манифестировать в любом возрасте, проявляется быстро прогрессирующим метаболическим кризом после длительного голодания или физической нагрузки. Тошнота, рвота, часто при нормальном уровне глюкозы в крови. При отсутствии терапии может привести к судорогам, коме и остановке сердца.
— Множественный дефицит ацил-КоA дегидрогеназ — глутаровая ацидурия тип II (ген ETFA)
Клинические проявления очень разнообразны: пороки развития головного мозга, кистозная болезнь почек, гипогликемия, ацидоз, эпилепсия, кардимиопатия.
— Митохондриального трифункционального белка дефицит (MTP) длинноцепочечной гидроксил-КоA дегидрогеназы дефицит (LCHAD) (ген HADHA)
Клинические проявления: ранняя манифестация, может включать кардиомипатию, нарушение работы печени, мышечную слабость, нейропатию, ретинопатию.
— Очень длинноцепочечной ацил-КоA дегидрогеназы дефицит VLCAD (ген ACADVL)
Клинические проявления: данное заболевание варьирует от умеренных, слабо выраженных симптомов у одних, до более серьезных проблем у других. Первые симптомы могут проявиться как с рождения, так и уже во взрослой жизни.
Младенческая форма — более тяжелая, протекает как LCHAD, манифестирует между рождением и 4 месяцами, может включать кардиомипатию, нарушение работы печени, мышечную слабость, нейропатию, ретинопатию
Детская форма — симптомы обычно появляются в раннем детском возрасте, обычно провоцируются болезнью или периодом длительного голодания. Проявляются симптомами гипогликемии и метаболическим кризом. Так же может включать следующие признаки: гепатомегалию, мышечную слабость, особенно после выраженной физической нагрузки. Патологии со стороны сердца при данном типе обычно не наблюдается. У некоторых детей симптомы данного заболевания выражены настолько слабо, что распознаются только после постановки диагноза брату или сестре.
Поздняя (взрослая) форма — клинические проявления: рабдомиолиз. Первые симптомы начинают проявляться в подростковом или уже во взрослом возрасте. Появляется общая мышечная слабость, которая становится продолжительной, более выраженная после физических нагрузок. Мышечная слабость сопровождается болезненностью, судорогами. Может появиться красновато-коричневый цвет мочи.
При данном типе, без лечения может развиться почечная недостаточность. Однако проблемы с сердцем и метаболический криз не характерны для данной формы и уровень глюкозы в крови остается относительно стабильным.
— HMG-КоA синтазы дефицит (ген HMGCS2)
Клинические проявления: острая гипокетотическая гипогликемия, относительно низкая переносимость голодания.
— HMG-КоA лиазы дефицит (ген HMGCL)
Клинические проявления: острая гипокетотическая гипогликемия, метаболический ацидоз, поражение печени.
ГЕНЕТИЧЕСКИЕ ПРИЧИНЫ НАРУШЕНИЯ ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
Гены представляют собой последовательность ДНК и в них записаны инструкции по «приготовлению» белков и РНК. Гены находятся в хромосомах. У человека 23 пары хромосом, одну из хромосом с соответствующим набором генов он наследует от матери, вторую от отца. Еще есть две половые хромосомы, которые определяют пол ребенка — у девочек две X-хромосомы, а у мальчиков одна X и одна Y-хромосомы.
Все заболевания из данной группы наследуются по аутосомно-рецессивному типу. Это значит, что оба родителя являются носителями мутации, но не болеют, т.к. у них есть вторая копия здорового гена. Риск рождения больного ребенка в данном случае составляет 25%. Больной ребенок наследует одну копию «больного» гена от отца и одну — от матери. Заболевание проявляется только в случае наличия двух копий поврежденного гена. Мальчики и девочки болеют с одинаковой частотой.
Рис. 4. Схема аутосомно-рецессивного типа наследования
ДИАГНОСТИКА НАРУШЕНИЙ ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
Основными методами диагностики этой группы болезней являются биохимические тесты. Проводят определение ацилкарнитинов в крови методом тандемной масс-спектрометрии.
При разных формах заболеваний наблюдается повышение определенных ацилкарнитинов.
Ацилкарнитины обозначают буквами с цифрой, которая указывает на число атомов углерода в данном соединении, другие обозначения относятся к наличию двойных связей, гидроксильной группы и т.д. Также разработаны методы определения активности ферментов, но их проводят очень небольшое число лабораторий во всем мире и их следует рекомендовать, если в результате ДНК-диагностики не удалось обнаружить мутации в гене.
Таблица 1. Соответствие между некоторыми FAOD и изменениями уровней ацилкарнитинов
ДНК-ДИАГНОСТИКА
Выявить мутацию (поломку) в гене можно молекулярно-генетическим методом, для этого проводится ДНК-диагностика. Однако, бывают случаи, когда мутацию найти с применением стандартных методов не удается. Тогда врачи при постановке диагноза ориентируются на клинические симптомы и биохимические изменения.
Если мутации найдены это дает возможность определить носителей в семье и провести пренатальную и/или преимплантационную диагностику.
МОГУТ ЛИ ДРУГИЕ ЧЛЕНЫ СЕМЬИ ЗАБОЛЕТЬ ИЛИ БЫТЬ НОСИТЕЛЯМИ?
Братья и сестры больного ребенка, могут быть больными, даже если у них на момент диагностики членов семьи не было симптомов. В данном случае необходимо провести диагностику, чтобы как можно раньше начать терапию и избежать осложнений. Также они могут быть носителями, как их родители. Что касается других членов семьи, то им важно сообщить, что они могут быть носителями. Это значит, что и у них есть риск рождения ребенка с данным заболеванием.
Если в семье уже есть один ребенок с нарушением окисления жирных кислот, когда у него рождается брат или сестра, нужно провести немедленную диагностику, на наличие данного заболевания, чтобы начать своевременную терапию.
ОСОБЕННОСТИ ТЕЧЕНИЯ ЗАБОЛЕВАНИЙ
Заболевания из этой группы могут начинаться в любом возрасте — с первых дней и даже часов жизни, в первые месяцы или проявиться только в детском возрасте или у подростков. Все зависит от формы болезни, тяжести мутации и внешних факторов (как часто ребенка кормят, как часто он болеет и т.д.).
Для некоторых форм более характерно поражение мышечной системы, для других — сердечной мышцы. Некоторые заболевания протекают преимущественно с поражением печени и с развитием метаболических кризов с гипогликемией.
Метаболические кризы — очень опасны. Именно в этот период могут развиться необратимые повреждения головного мозга, который крайне нуждается в поступлении метаболической энергии в виде глюкозы или кетоновых тел. Если при многих других наследственных нарушениях обмена веществ, сопровождающихся гипогликемией (например, нарушениях обмена гликогена) наблюдается высокий уровень кетоновых тел и мозг может получать энергию за счет их метаболизма, при нарушениях окисления жирных кислот уровень кетоновых тел низкий, поэтому повреждение головного мозга развивается очень быстро.
ОБРАЗ ЖИЗНИ, ЕЖЕДНЕВНАЯ ТЕРАПИЯ И ДИЕТА
Долгосрочная терапия в данной группе заболеваний — это диетотерапия.
Цели:
— Нормальное физическое и интеллектуальное развитие.
— Прибавка в весе и росте, в соответствие с возрастом.
— Предупреждение характерных симптомов, например мышечной боли, мышечной слабости.
— Предотвращение опасных проявлений, таких как повреждение сердечной мышцы.
— Предупреждение катаболических ситуаций, которые могут привести к метаболическим кризам с гипогликемией и разрушению мышечных клеток.
Для достижения этих целей диетотерапия должна отвечать следующим требованиям:
— Обеспечивать адекватное потребление белка в соответствие с возрастом и потребностью.
— Обеспечивать незаменимыми жирными кислотами, витаминами и минералами.
— покрывать энергозатраты.
— Избегать длительных периодов голодания
— Ограничивать потребление жирных кислот с длинной цепью.
— Соответствовать вкусу, возрасту и потребностям ребенка.
— Подходить для повседневной жизни.
Заболевания, при которых диета не используется:
— Первичный дефицит карнитина
— Короткоцепочечной ацил-КоA дегидрогеназы дефицит (SCAD)
— Среднецепочечной ацил-КоA дегидрогеназы дефицит MCAD
Заболевания, при которых используется диетотерапия:
— Карнитин пальмитоилтрансферазы 1 дефицит (CPTI)
— Карнитин пальмитоилтрансферазы 2 дефицит (CPTII)
— Митохондриального трифункционального белка дефицит (MTP) длинноцепочечной гидроксил-КоA дегидрогеназы дефицит (LCHAD)
— Очень длинноцепочечной ацил-КоA дегидрогеназы дефицит (VLCAD)
— Множественный дефицит ацил-КоA дегидрогеназ
— HMG-КоA синтазы дефицит
— HMG-КоA лиазы дефицит
1. Умеренное ограничение жира в рационе:
— Карнитин пальмитоилтрансферазы 1 дефицит
2. Умеренное или значительное снижение жира в рационе:
— Карнитин пальмитоилтрансферазы 2 дефицит
— Очень длинноцепочечной ацил-КоA дегидрогеназы дефицит VLCAD (бессимптомная форма)
— Множественный дефицит ацил-КоA дегидрогеназ
3. Значительное снижение жира с использованием MCT (среднецепочечных триглицеридов) ночные кормления:
— Митохондриального трифункционального белка дефицит (MTP) длинноцепочечной гидроксил-КоA дегидрогеназы дефицит (LCHAD)
— Очень длинноцепочечной ацил-КоA дегидрогеназы дефицит VLCAD
Таблица 2. Максимальный период голода в зависимости от возраста:
Внимание! В таблице приведены усредненные интервалы между приемом пищи. Интервалы между приемами пищи в дневные часы и ночные перерывы между кормлениями устанавливаются индивидуально, в зависимости от уровня гликемии.
Таблица 3. Среднесуточные нормы физиологических потребностей в энергии для детей согласно FAO/WHO/UNU 2001 (WHO Technical Report Series 17 — 24 okt 2001)
Возраст (мес.) | Энергия (ккал/кг) муж | Энергия (ккал/кг) жен |
0 — 1 | 113 | 107 |
1 — 2 | 104 | 101 |
2 — 3 | 95 | 94 |
3 — 4 | 82 | 84 |
4 — 5 | 81 | 83 |
5 — 6 | 81 | 82 |
6 — 7 | 79 | 78 |
7 — 8 | 79 | 78 |
8 — 9 | 79 | 78 |
9 — 10 | 80 | 79 |
10 — 11 | 80 | 79 |
11 — 12 | 81 | 79 |
Возраст (г.) | ||
1 — 2 | 82,4 | 80,1 |
2 — 3 | 83,6 | 80,6 |
3 — 4 | 79,7 | 76,5 |
4 — 5 | 76,8 | 73,9 |
5 — 6 | 74,5 | 71,5 |
6 — 7 | 72,6 | 69,3 |
7 — 8 | 70,5 | 66,7 |
8 — 9 | 68,5 | 63,8 |
9 — 10 | 66,6 | 60,8 |
10 — 11 | 64,6 | 57,8 |
11 — 12 | 62,4 | 54,8 |
12 — 13 | 60,2 | 52,0 |
13 — 14 | 57,9 | 49,3 |
14 — 15 | 55,6 | 47,0 |
15 — 16 | 53,4 | 45,3 |
16 — 17 | 51,6 | 44,4 |
17 — 18 | 50,3 | 44,1 |
Диетотерапия при бессимптомной форме недостаточности очень длинноцепочечной ацил-КоA дегидрогеназы (VLCAD):
— 0 — 4 мес. — 50% грудного молока 50% специализированное питание с MCT
— > 4 мес. — умеренное сокращение жира до 30% энергии от общего калоража, из них 10 — 15% за счет MCT, 3 — 4% за счет незаменимых жирных кислот.
Диетотерапия при недостаточности очень длинноцепочечной ацил-КоA дегидрогеназы (VLCAD) с ранней манифестацией и недостаточности митохондриального трифункционального белка (MTP) недостаточности длинноцепочечной гидроксил-КоA дегидрогеназы (LCHAD):
— 0 — 4 мес. — 100% специализированное питание с MCT
— > 4 мес. — сокращение жира до 25 — 30% энергии от общего калоража, 20 — 25% из них за счет MCT, 3 — 4% за счет незаменимых жирных кислот. Снизить потребление длинноцепочечных жирных кислот максимально на сколько возможно.
Для всех типов: избегать катаболических ситуаций!!!
ПРИМЕР РАСЧЕТА ДИЕТЫ
Расчет диеты для ребенка с диагнозом: недостаточность длинноцепочечной гидроксил-КоA дегидрогеназы (LCHAD):
Возраст 1,5 года, вес 10 кг.
Энергия:
В 1,5 года суточная калорийность рациона составляет 90 ккал/кг/сут, следовательно, ваш ребенок должен получить 10 x 90 = 900 ккал в сутки.
Жиры:
25% энергии должны обеспечивать жиры.
1 г жира — это 9 калорий (Ккал)
Исходя из этого, ребенок должен получить:
Жиры (г/сут) = 25% x 900 / 9 / 100% = 25 г жира в сутки.
20% энергии должны обеспечивать жирные кислоты со средней цепью, следовательно:
MCT (г/сут) = 20% x 900 / 9 / 100% = 20 г MCT в сутки.
Получается, что на долю жиров с длинной цепью приходится только: 25 г — 20 г = 5 г в сутки.
3 — 4% энергии мы должны обеспечить за счет незаменимых жирных кислот (ПНЖК).
Считаем:
ПНЖК (г/сут) = 3% x 900 / 9 / 100% = 3 г в сутки
Белки:
1 г белка — это 4 калорий (Ккал)
При недостаточности очень длинноцепочечной ацил-КоA дегидрогеназы (VLCAD) с ранней манифестацией и недостаточности длинноцепочечной гидроксил-КоA дегидрогеназы (LCHAD) более целесообразна диета с повышенным содержанием белка 1,5 — 2,5 г/кг/сут.
В приведенном выше примере ребенку весом 10 кг суточная потребность в белке составляет: 15 — 25 г
Белок 2 г/кг/сут — это 20 г, в процентах от общего калоража это:
Белки (%) = 4 x 20 / 900 x 100% = 9%
Углеводы:
1 г углеводов — это 4 калорий (Ккал)
И так мы посчитали, что суточная калорийность рациона у нашего ребенка 900 ккал, из них 25% энергии обеспечивают жиры, 9% белки, оставшуюся энергию мы должны покрыть за счет углеводов:
100% — 25% — 8% = 66%
Переводим в граммы:
Углеводы (г/сут) = 66% x 900 / 4 / 100% = 148 г
ПОЛИНЕНАСЫЩЕННЫЕ НЕЗАМЕНИМЫЕ ЖИРНЫЕ КИСЛОТЫ (ПНЖК)
Существует 2 вида ПНЖК: омега-3, омега-6.
ПНЖК занимают большую часть состава защитной мембраны (оболочки) любой клетки. Организм способен преобразовывать жирные кислоты одного класса в другой, но не способен синтезировать оба класса из более простых веществ, поэтому они обязательно должны присутствовать в пище, подобно микроэлементам.
В нашем организме ПНЖК имеют свойства в этих же оболочках обновляться по мере того, как клетка проходит свой естественный жизненный цикл.
Организм всегда использует те жирные кислоты, которые есть в наличии. Именно поэтому целесообразно, включить в рацион ПНЖК класса омега-3 и омега-6 в правильном соотношение.
Омега-3: АЛК (Альфа-линоленовая кислота), ДГК (докозагексаеновая кислота) и ЭПК (эйкозапентаеновая кислота).
Омега-6: линолевая кислота, арахидоновая кислота.
Таблица 4. ПНЖК при низкожировой диете, оптимальное соотношение:
Возраст | Омега-6 (% от энергии) | Омега-3 (% от энергии) | Общая сумма (% от энергии) | Омега-6: Омега-3 | Омега-6 (г/сут) | Омега-3 (г/сут) |
0 — < 4 мес. | 4,0 | 0,5 | 4,5 | 8:1 | 2,0 | 0,25 |
4 — 12 мес. | 3,5 | 0,5 | 4,0 | 7:1 | 2,7 | 0,4 |
1 — < 4 л. | 3,0 | 0,5 | 3,5 | 6,5:1 | 3,5 | 0,6 |
> 4 л. | 2,5 | 0,5 | 3,0 | 5:1 | 5,5 | 1,1 |
Таблица 5. Содержание ПНЖК в растительных маслах:
масла | Линоленовая кислота (омега-6) | Альфа-линоленовая кислота (омега-3) | Омега-6: омега-3 |
Сафлоровое масло | 75 | 0,47 | 150:1 |
Тыквенное масло | 49 | 0,5 | 98:1 |
Льняное масло | 14 | 53 | 0,26:1 |
Кукурузное масло | 55,5 | 1,0 | 55:1 |
Оливковое масло | 8,2 | 0,86 | 10:1 |
Рапсовое масло | 15 | 8,6 | 1,7:1 |
Кунжутное масло | 42,5 | 0,8 | 53:1 |
Масло соевых бобов | 53 | 7,7 | 7:1 |
Масло подсолнечное | 50,1 | 0,2 | 62:1 |
Масло грецкого ореха | 52,4 | 12,2 | 4:1 |
Масло зародышей пшеницы | 55,7 | 7,8 | 7:1 |
Докозагексаеновая кислота
Учитывая, что лишь малая часть ДГК кислоты синтезируется из альфа-линоленовой, одних растительных источников для восполнения необходимого уровня Омега-3 недостаточно. Именно поэтому в рацион рекомендуется включить добавки содержащие докозогексаеновую кислоту (ДГК) в необходимом количестве. Доза ПНЖК определяется врачом.
КАРНИТИН
При первичном дефиците карнитина, препараты левокарнитина назначают в высоких дозах 50 — 400 мг/кг/сут.
В остальных случаях карнитин назначают в небольших дозировках с осторожностью per.os. (доза определяется врачом)
ПРОТИВОПОКАЗАНО внутривенное введение раствора карнитина при нарушении b-окисления жирных кислот!!!
КАКИМ ОБРАЗОМ ПИТАНИЕ РЕБЕНКА МОЖЕТ БЫТЬ АДАПТИРОВАНО К ПИТАНИЮ СЕМЬИ
К концу первого года жизни ребенок начинает переходить на общий стол, его питание все больше начинает походить на взрослую пищу. Еда имеет более твердую консистенцию, и ассортимент отдельных продуктов и блюд расширяется.
Рис. 1. Пищевая пирамида, иллюстрирующая состав ежедневного рациона ребенка с расстройством метаболизма длинноцепочечных жирных кислот.
Большая часть продуктов соответствует рекомендациям для здорового, разнообразного питания детей со здоровым метаболизмом. Очень важны напитки без сахара, такие как минеральная вода, фруктовый чай и небольшое количество фруктового сока, смешанного с газированной водой. В приоритете фрукты и овощи. В их составе практически не содержится жиров, поэтому они могут употребляться в большом количестве. Рекомендуется, как и всем, съедать 5 порций фруктов и овощей в день.
Следующий уровень пищевой пирамиды содержит зерновые продукты, злаки и картофель. При их приготовлении должно использоваться минимальное количество жира.
Обезжиренное молоко и молочные продукты обогащают ежедневный рацион и дополняются меньшим количеством нежирных сортов мяса и рыбы
Масла и твердые жиры должны быть заменены MCT и незаменимыми жирными кислотами в правильном соотношении.
На вершине пирамиды находятся низкожировые сладости.
МЕТАБОЛИЧЕСКИЙ КРИЗ
Метаболический криз — критическое, угрожающее жизни состояние, спровоцированное неблагоприятными факторами, обуславливающими усиленный распад жиров с образованием жирных кислот, нарушением их окисления и приводящее к образованию токсичных метаболитов.
Что может спровоцировать метаболический криз:
— длительное голодание
— инфекция
— тяжелая физическая и эмоциональная нагрузка
Признаки гипогликемии (снижение уровня сахара в крови):
— Слабость
— Дрожь
— Головокружение
— Липкий холодный пот.
Первые симптомы метаболического криза:
Чрезвычайная сонливость, изменение поведения, раздражительность, снижение аппетита. Часто криз провоцирует инфекция, поэтому первые проявления могут начинаться лихорадкой, тошнотой, диареей, рвотой. Затем следует снижение сахара в крови. Если своевременно не начать лечение у ребенка может появиться одышка, судороги и кома.
В КАКИХ СЛУЧАЯХ НУЖНО НЕМЕДЛЕННО ОБРАТИТЬСЯ ЗА МЕДИЦИНСКОЙ ПОМОЩЬЮ
Если вы заметили любой из этих симптомов у вашего ребенка:
— Плохой аппетит
— Выраженная слабость или чрезмерная сонливость
— Рвота
— Диарея
— Лихорадка
— Постоянные мышечные боли, слабость или красновато-коричневый цвет мочи.
ЧТО ДЕЛАТЬ ЕСЛИ ВЫ ЗАМЕТИЛИ У РЕБЕНКА ЭТИ СИМПТОМЫ?
Ухудшение состояния обычно наблюдается в тот момент, когда организму не хватает энергии. И он начинает ее искать всеми возможными способами, один из путей — это распад собственных тканей (катаболизм). Допускать этого ни в коем случае нельзя. И первое, что мы должны сделать, это попробовать восполнить энергию за счет глюкозы. Дайте ребенку углеводы, это может быть фруктовый сок, сладкий чай, или раствор глюкозы.
Учитывая высокий риск развития метаболического криза при появлении вышеописанных симптомов нужно немедленно обратиться за медицинской помощью. Необходимо взять анализ крови на электролиты (K , NA , Ca ), глюкозу, КЩС, АлАТ, АсАТ, КФК, ЛДГ, ХС, ЛПВП, ЛПНП, триглицериды, NH3. И начать внутривенное введение глюкозы, согласно протоколу:
При невозможности организации питания через рот необходимо установить назогастральный зонд
ТЕРАПИЯ
Принципы терапии:
— Избегать голодания
— Сокращение в рационе жирных кислот с длинной цепью
— Обогащение пищи среднецепочечными жирными кислотами
— Предотвратить гипогликемию
— Дополнительная метаболическая поддержка при развитии инфекционных заболеваний
— Добавки карнитина (см. раздел карнитин)
ФИЗИЧЕСКАЯ НАГРУЗКА
Физическая нагрузка — это всегда дополнительная энергия, те калории, которые мы должны заплатить нашему организму за усиленную работу.
Поэтому если вы знаете, что собираетесь потратить больше энергии чем обычно, вы должны быть к этому готовы.
За 30 минут до физической нагрузки рекомендовано принять 0,2 — 0,5 г MCT на кг массы тела. Для большей эффективности можно смешать с углеводами, например, яблочным соком.
После физической нагрузки нужно восполнить потраченную энергию белково-углеводной закуской, отношение углеводов к белкам — 3:1
Специализированное питание
Для пациентов с нарушениями окисления жирных кислот разработаны несколько вариантов специализированного питания и отдельные лечебные продукты. Они отличаются между собой. При переходе с одной формулы на другую и с одного продукта на другой это нужно учитывать!
Таблица 7. Состав специализированного питания при FAOD
ДИСПАНСЕРИЗАЦИЯ
Для пациентов с нарушениями окисления жирных кислот необходимы регулярные обследования. В таблице приведен примерный перечень и регулярность исследований, но в каждом случае вы должны согласовывать план обследования со своим лечащим врачом.
Таблица 8. Регулярность различных исследований при FAOD
ТЕЧЕНИЕ БЕРЕМЕННОСТИ ПРИ НАРУШЕНИИ В-ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ У ПЛОДА
При беременности при некоторых формах болезней есть высокие риски развития осложнений. Среди них чаще всего встречаются: сильный токсикоз, частая рвота, желтуха, боль в животе, повышение артериального давления, кровотечение.
ПРОГНОЗ ПРИ СВОЕВРЕМЕННОЙ ПОСТАНОВКИ ДИАГНОЗА
Прогноз зависит от формы заболевания и манифестации. В большинстве случаев при своевременной диагностики и терапии дети с заболеванием могут рассчитывать на полноценную жизнь, ничем с первого взгляда не отличающеюся от остальных. Однако терапия будет пожизненной, и результат будет зависеть от правильности соблюдений рекомендаций врачей.
ДОПОЛНИТЕЛЬНАЯ ИНФОРМАЦИЯ
Печатая в поисковике термин «fatty acids oxidation defects», вы найдете тысячи английских ссылок. В русскоязычном интернете полезных сайтов на эту тему пока нет. Эти сайты создают сами родители, при поддержке врачей и специалистов. Поэтому если у вас есть силы и желание — сделайте такой сайт для других семей и для себя.
Интернет приносит большую пользу, но иногда информация, найденная в интернете может не соответствовать рекомендациям, которым вы следуете. Не бросайтесь выполнять то, что написано на страницах соцсетей и на сайтах, даже если информация заслуживает доверия! Обсудите это с вашим лечащим врачом, вы должны быть уверены, что делаете все правильно.
При составлении данной брошюры мы пользовались следующими рекомендациями и источниками:
Treatment recommendations in long-chain fatty acid oxidation defects: consensus from a workshop — Spiekerkoetter U, Lindner M, Santer R, Grotzke M, Baumgartner MR, Boehles H, Das A, Haase C, Hennermann JB, Karall D, de Klerk H, Knerr I, Koch HG, Plecko B, Roschinger W, Schwab KO, Scheible D, Wijburg FA, Zschocke J, Mayatepek E, Wendel U. (Department of General Pediatrics, University Children’s Hospital, Dusseldorf, Germany) 2009, Apr 28.
Vademecum Metabolicum — Johannes Zschoke (Innsbruck, Austria), Georg F. Hoffmann, Heidelberg, Germany)
http://www.fodsupport.org
СОКРАЩЕНИЯ И ПОНЯТИЯ
— CPT1 — Карнитин пальмитоилтрансферазы 1 дефицит
— CPT2 — Карнитин пальмитоилтрансферазы 2 дефицит
— SCAD — Короткоцепочечной ацил-КоA дегидрогеназы дефицит
— MCAD — Среднецепочечной ацил-КоA дегидрогеназы дефицит
— MTP — Митохондриального трифункционального белка дефицит
— LCHAD — длинноцепочечной гидроксил-КоA дегидрогеназы дефицит
— VLCAD — Очень длинноцепочечной ацил-КоA дегидрогеназы дефицит
— СЦТ (MCT) — среднецепочечные жирные кислоты (триглицериды)
— ДЦТ — длинноцепочечные жирные кислоты (триглицериды)
— ПНЖК — полиненасыщенные незаменимые жирные кислоты
— АЦИЛКАРНИТИНЫ — жирные кислоты связанные с карнитином
— КАРДИОМИОПАТИЯ — заболевание сердца, при которых сердечная мышца структурно и функционально изменена
— ГИПОТОНИЯ — мышечная слабость
— ГИПОГЛИКЕМИЯ — снижение уровня сахара в крови
— РАБДОМИОЛИЗ — разрушение клеток мышечной ткани
— МЕТАБОЛИЧЕСКИЙ АЦИДОЗ — нарушение кислотно-основного состояния, проявляющееся низкими значениями pH крови и низкой концентрацией бикарбоната в крови
— ГИПОГЛИКЕМИЯ — снижение уровня сахара в крови
— ГИПЕРАММОНИЕМИЯ — повышение уровня аммония в крови
— НЕЙРОПАТИЯ — поражение нерва, при котором происходит нарушение его целостности или целостности его оболочки.
— РЕТИНОПАТИЯ — поражение сетчатки
— КАТАБОЛИЗМ — процесс распада сложных веществ.
Приложение Г1 — ГN
ШКАЛЫ ОЦЕНКИ, ВОПРОСНИКИ И ДРУГИЕ ОЦЕНОЧНЫЕ ИНСТРУМЕНТЫ
СОСТОЯНИЯ ПАЦИЕНТА, ПРИВЕДЕННЫЕ В КЛИНИЧЕСКИХ РЕКОМЕНДАЦИЯХ
Приложение Г1
КЛАССИФИКАЦИЯ НАСЛЕДСТВЕННЫХ НАРУШЕНИЙ ОКИСЛЕНИЯ
ЖИРНЫХ КИСЛОТ
Приложение Г2
КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ НАРУШЕНИЙ ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
Клинические признаки | Нарушения |
Энцефалопатия (судороги, летаргия, кома) | Все формы, кроме мышечного типа недостаточности карнитина |
Поражение печени | Все формы |
Мышечная слабость, гипотония | Все формы |
Миалгия, рабдомиолиз, изменение цвета мочи (красновато-бурая) | CPT2, CACT, VLCAD, TFP, LCHAD, ГА2 |
Кардиомиопатия | Карнитина недостаточность, CPT1 (редко), CPT2, CACT, VLCAD, TFP, LCHAD, ГА2 |
Нарушения ритма сердца | CPT2, CACT, VLCAD, TFP, LCHAD, MCAD, ГА2 |
Синдром внезапной детской смерти | Карнитина недостаточность, CPT2, CACT, VLCAD, TFP, LCHAD, MCAD, ГА2 |
Лицевые дизморфии | CPT2, ГА2 |
Тубулярные нарушения | CPT1, CPT2, LCHAD, ГА2 |
Почечная недостаточность | CPT1, CPT2, CACT, VLCAD, TFP, LCHAD, ГА2 |
Наличие кист в почках | CPT2, ГА2 |
Увеличение почек | CPT1, CPT2, ГА2 |
Боли в животе, диарея | Карнитина недостаточность, CPT1, TFP, LCHAD |
Периферическая нейропатия | TFP, LCHAD |
Пигментный ретинит, помутнение хрусталика (редко) | TFP, LCHAD |
Необычный запах мочи (типа «потных ног») | ГА2 |
Осложнения беременности у матерей (неукротимая рвота, увеличение печени, повышение активности трансаминаз, гипербилирубинемия, жировая дистрофия печени) | CPT1, TFP, LCHAD |
Приложение Г2а
ОБЩИЕ КЛИНИЧЕСКИЕ ПРОЯВЛЕНИЯ FAOD
Приложение Г3
ЛАБОРАТОРНЫЕ ИЗМЕНЕНИЯ ПРИ НАРУШЕНИЯХ ОКИСЛЕНИЯ
ЖИРНЫХ КИСЛОТ
Заболевание | Лабораторные изменения |
Первичный дефицит карнитина | Кровь: гипокетотическая гипогликемия на фоне голода, КФК, АСТ, АЛТ Моча: Дикарбоновые кислоты в норме или несколько повышены Ацилкарнитины: :C0 < 10, суммарные ацилкарнитины < 5 |
Карнитин пальмитоилтрансферазы 1A дефицит (CPT1) | Кровь: гипокетотическая гипогликемия на фоне голода, КФК, АСТ, АЛТ NH3; Коагулопатия; Моча: отсутствие повышения дикарбоновых кислот Ацилкарнитины: C0, C0 / (C16 C18) C16, C18, C18:1 |
Карнитин-ацилкарнитин транслоказы дефицит (CACT) | Кровь: гипокетотическая гипогликемия на фоне голода, КФК, АСТ, АЛТ, лактат, NH3 Коагулопатия Моча: может отмечаться повышение дикарбоновых кислот Ацилкарнитины: C16, C16:1, C18, C18:1 C0 |
Карнитин пальмитоилтрансферазы 2 дефицит (CPT2) | Кровь: гипокетотическая гипогликемия на фоне голода КФК, АСТ, АЛТ Моча: миоглобинурия могут повышаться дикарбоновые кислоты Ацилкарнитины: C16, C16:1, C18, C18:1 C0 |
Короткоцепочечной ацил-КоA дегидрогеназы дефицит (SCAD) | Кровь: метаболический ацидоз, гипогликемия Моча: этилмалоновая кислота Ацилкарнитины: C4 |
Среднецепочечной ацил-КоA дегидрогеназы дефицит (MCAD) | Кровь: гипокетотическая гипогликемия на фоне голода, АСТ, АЛТ АЛТ Моча: C6 — C10 дикарбоновые кислоты, суберилглицин, гексаноилглицин Ацилкарнитины: C8, C10, C10:1 C0 (или норма) |
Очень длинноцепочечной ацил-КоA дегидрогеназы дефицит (VLCAD) | Кровь: гипокетотическая гипогликемия на фоне голода, КФК, АСТ, АЛТ, NH3 Моча: C6 — C14 дикарбоновые кислоты Ацилкарнитины: C12:1, C14, C14:1, C16, C16:1 |
Короткоцепочечной 3-гидроксиацил-КоA дегидрогеназы дефицит (SCHAD) | Кровь: гипокетотическая гипогликемия на фоне голода, инсулин, NH3 Моча: 3-гидроксиглутаровая кислота Ацилкарнитины: C4OH |
Длинноцепочечной 3-гидроксиацил-КоA дегидрогеназы дефицит (LCHAD) | Кровь: гипокетотическая гипогликемия на фоне голода, КФК, АСТ, АЛТ, NH3 Моча: C6 — C14 дикарбоновые кислоты; Ацилкарнитины: :C16:1-OH, C16-H, C18:1-OH, C18OH |
Митохондриального трифункционального белка дефицит (TFP) | Кровь: гипокетотическая гипогликемия на фоне голода, КФК, АСТ, АЛТ, NH3 Моча: C6 — C14 дикарбоновые кислоты; Ацилкарнитины: :C16:1-OH, C16-H, C18:1-OH, C18OH |
Множественный дефицит ацил-КоA дегидрогеназ дефицит глутаровая ацидурия тип II (ГА2 ГА2 ГА2 ГА2) | Сыворотка: гипокетотическая гипогликемия на фоне голода, гипокетотическая гипогликемия на фоне голода, КФК, АСТ, АЛТ, NH3, метаболический ацидоз Моча: C5 — C10 дикарбоновые кислоты, молочная, глутаровая, этилмолоновая кислоты. Ацилкарнитины: C4 — C18 |
Приложение Г4
ЗАБОР БИОМАТЕРИАЛА ДЛЯ ДИАГНОСТИКИ В ПЯТНАХ КРОВИ
Кровь собирается на стандартную карточку-фильтр (N903), которая применяется для скрининга новорожденных в Российской Федерации или аналогичную для получения сухого пятна крови. Кровь может быть, как капиллярная (из пальца, пятки), так и венозная. Венозная кровь собирается в пробирку, аккуратно перемешивается и затем быстро с помощью пипетки наносится на фильтр по 25 — 50 мкл крови на каждую выделенную область. Необходимо хорошо пропитать выделенную область на фильтре (рис. 1). Предпочтительно собирать образцы после еды через 40 минут — 1 час. Возможно также осуществить забор крови и натощак. На карточке-фильтре обязательно должны быть четко указаны ФИО, кем и откуда направлен пациент, дата рождения и телефон лечащего врача (рисунок 2). Образец сухого пятна крови вкладывается в чистый конверт, либо в чистый файл. Карточка-фильтр не должна соприкасаться с грязной поверхностью и с образцами других пациентов. Необходимо приложить к образцам информированные согласия пациента или его законных представителей на проведение лабораторных исследований | |
— Капли должны полностью пропитать кружки с обратной стороны — Не касайтесь области, пропитанной кровью | |
Рисунок 1. Образец правильного нанесения крови на карточку-фильтр | |
Рисунок 2. Образец карточки-фильтра |
Алгоритм действий медицинского персонала при взятии образцов крови
— вымыть руки (гигиенический уровень), надеть перчатки;
— вымыть руки пациента (пятку ребенка, в случае, если кровь берется из пятки);
— протереть область прокалывания стерильной салфеткой, смоченной 70% спиртом, промокнуть сухой стерильной салфеткой; — проколоть стерильным одноразовым скарификатором;
— снять первую каплю крови стерильным сухим тампоном из нетканого материала или марли или салфеткой марлевой медицинской стерильной;
— мягко надавить для получения второй капли крови;
— приложить перпендикулярно тест-бланк к капле крови и пропитать его кровью насквозь;
— аналогичным образом нанести на тест-бланк 6 — 8 капель, вид пятен крови должен быть одинаковым с обеих сторон.
— высушить тест-бланк в горизонтальном положении на чистой обезжиренной поверхности не менее 4 ч без применения тепловой обработки и попадания прямых солнечных лучей;
— упаковать тест-бланки в чистый конверт таким образом, чтобы пятна крови не соприкасались.
Особенности при инфузионной терапии
Некоторые пациенты могут получать инфузионную терапию, переливание компонентов крови, что может оказать влияние на результаты тестов. Например, при переливании плазмы крови могут быть получены ложноотрицательные результаты. Рекомендуется осуществить забор крови для диагностики не ранее чем через 6 — 7 дней после переливания плазмы крови и через 7 — 10 дней после переливания компонентов крови
Не допускается забор крови
— сразу после проведения пациенту инфузионной терапии;
— сразу после заменного переливания крови.
Хранение и транспортировка биоматериала
Образцы высушенных пятен крови можно хранить в обычной камере холодильника при 4 °C до отправки. Срок хранения до момента отправки не должен превышать 7 дней. Если хранить дольше и при более высокой температуре, то это приведет к ложноположительным результатам.
Приложение Г5
МАКСИМАЛЬНАЯ ПРОДОЛЖИТЕЛЬНОСТЬ ГОЛОДАНИЯ (ПРИ СТАБИЛЬНЫХ
МЕТАБОЛИЧЕСКИХ СОСТОЯНИЯХ)
Дефицит среднецепочечной ацил-КоA дегидрогеназы (MCAD): рекомендации по максимальному безопасному времени голодания для здорового ребенка.
———————————
<*> Возраст около 2 недель.
Источник: www.bimdg.org
Приложение Г6
СРЕДНЕСУТОЧНЫЕ НОРМЫ ФИЗИОЛОГИЧЕСКИХ ПОТРЕБНОСТЕЙ
В ЭНЕРГИИ ДЛЯ ДЕТЕЙ
Среднесуточные нормы физиологических потребностей в энергии для детей согласно FAO/WHO/UNU 2001 (WHO Technical Report Series 17 — 24 okt 2001) | ||
Возраст (мес.) | Энергия (ккал/кг) муж. | Энергия (ккал/кг) жен. |
0 — 1 | 113 | 107 |
1 — 2 | 104 | 101 |
2 — 3 | 95 | 94 |
3 — 4 | 82 | 84 |
4 — 5 | 81 | 83 |
5 — 6 | 81 | 82 |
6 — 7 | 79 | 78 |
7 — 8 | 79 | 78 |
8 — 9 | 79 | 78 |
9 — 10 | 80 | 79 |
10 — 11 | 80 | 79 |
11 — 12 | 81 | 79 |
Возраст (г.) | ||
1 — 2 | 82,4 | 80,1 |
2 — 3 | 83,6 | 80,6 |
3 — 4 | 79,7 | 76,5 |
4 — 5 | 76,8 | 73,9 |
5 — 6 | 74,5 | 71,5 |
6 — 7 | 72,6 | 69,3 |
7 — 8 | 70,5 | 66,7 |
8 — 9 | 68,5 | 63,8 |
9 — 10 | 66,6 | 60,8 |
10 — 11 | 64,6 | 57,8 |
11 — 12 | 62,4 | 54,8 |
12 — 13 | 60,2 | 52,0 |
13 — 14 | 57,9 | 49,3 |
14 — 15 | 55,6 | 47,0 |
15 — 16 | 53,4 | 45,3 |
16 — 17 | 51,6 | 44,4 |
17 — 18 | 50,3 | 44,1 |
Приложение Г7
СООТНОШЕНИЕ ЖИРОВ И УГЛЕВОДОВ В ДИЕТЕ ПРИ РАЗНЫХ
ФОРМАХ FAOD
Заболевание (сокращенно) | Диета (доля от общей калорийности) |
OCTN2, MCAD ETF-B2 Мягкая, поздняя, мышечная, асимптотическая формы: CPT2, VLCAD, SCAD,, ETF/ETF-DH HMGSC2, OXCT | Нормальная: жиры: 30%; углеводы: 50 — 55%; белки: 10 — 15% |
CACT, CPT2, M/SCHAD, ETF/ETF-DH, MKAT | Высокоуглеводная, низкожировая: жиры: 20 — 25% (включая незаменимые жирные кислоты); углеводы: 65 — 75%; белки: 8 — 10%. |
CPT1, CACT, CPT2, VLCAD, LCHAD, FATP1 | Высокоуглеводная, низкожировая: жиры: 20 — 25% (включая: 10% ДЦТ, 10 — 15% MCT, 1 — 4% незаменимые жирные кислоты); углеводы: 65 — 75%; белки: 8 — 10%. |
Приложение Г8
ДОБАВКИ НЕЗАМЕНИМЫХ ЖИРНЫХ КИСЛОТ ПРИ ДИЕТЕ С НИЗКИМ
СОДЕРЖАНИЕМ ЖИРОВ (D-A-CH)
Возраст | % энергии | ||
n-6 линолевая кислота | n-3 линоленовая кислота | Суммарно | |
от 0 до < 4 месяцев | 4,0 | 0,5 | 4,5 |
от 4 до < 12 месяцев | 3,5 | 0,5 | 4,0 |
от 1 до < 4 лет | 3,0 | 0,5 | 3,5 |
> 4 лет | 2,5 | 0,5 | 3,0 |
Приложение Г9
ПРИМЕНЕНИЕ УГЛЕВОДОВ ПРИ ПОДОЗРЕНИИ НА МЕТАБОЛИЧЕСКИЙ КРИЗ
И ПРИ ИНТЕРКУРРЕНТНЫХ ЗАБОЛЕВАНИЯХ
[81]
Соответствие между возрастом ребенка и дозой глюкозы для энтерального применения при подозрении на метаболический криз.
[130]
Среднесуточные нормы физиологических потребностей в энергии для людей разных возрастов согласно FAO/WHO/UNU 2007 (WHO Technical Report Series 2007) | ||
Возраст (г.) | Энергия (ккал/кг) муж. | Энергия (ккал/кг) жен. |
0,5 | 80,0 | 81,2 |
2,5 | 83,1 | 79,8 |
5,0 | 75,2 | 72,8 |
10 | 65,7 | 59,2 |
15 | 54,9 | 46,1 |
Взрослые, ведущие образ жизни средней активности, вес 70 кг | ||
18 — 29 | 43,7 | 38,0 |
30 — 59 | 41,8 | 35,3 |
Взрослые, ведущие образ жизни средней активности, вес 50 кг | ||
18 — 29 | 50,6 | 43,0 |
30 — 59 | 50,6 | 43,7 |
Среднесуточные нормы физиологических потребностей в энергии для детей согласно FAO/WHO/UNU 2001 (WHO Technical Report Series 17 — 24 okt 2001) | ||
Возраст (мес.) | Энергия (ккал/кг) муж. | Энергия (ккал/кг) жен. |
0 — 1 | 113 | 107 |
1 — 2 | 104 | 101 |
2 — 3 | 95 | 94 |
3 — 4 | 82 | 84 |
4 — 5 | 81 | 83 |
5 — 6 | 81 | 82 |
6 — 7 | 79 | 78 |
7 — 8 | 79 | 78 |
8 — 9 | 79 | 78 |
9 — 10 | 80 | 79 |
10 — 11 | 80 | 79 |
11 — 12 | 81 | 79 |
Возраст (г.) | ||
1 — 2 | 82,4 | 80,1 |
2 — 3 | 83,6 | 80,6 |
3 — 4 | 79,7 | 76,5 |
4 — 5 | 76,8 | 73,9 |
5 — 6 | 74,5 | 71,5 |
6 — 7 | 72,6 | 69,3 |
7 — 8 | 70,5 | 66,7 |
8 — 9 | 68,5 | 63,8 |
9 — 10 | 66,6 | 60,8 |
10 — 11 | 64,6 | 57,8 |
11 — 12 | 62,4 | 54,8 |
12 — 13 | 60,2 | 52,0 |
13 — 14 | 57,9 | 49,3 |
14 — 15 | 55,6 | 47,0 |
15 — 16 | 53,4 | 45,3 |
16 — 17 | 51,6 | 44,4 |
17 — 18 | 50,3 | 44,1 |
Приложение Г10
СОДЕРЖАНИЕ MCT В СМЕСЯХ ДЛЯ ЛЕЧЕБНОГО ПИТАНИЯ И ПРОДУКТАХ
ДОПОЛНИТЕЛЬНОГО ПИТАНИЯ, ПРИМЕНЯЕМЫХ У ПАЦИЕНТОВ С НОЖК
[105]
Химический состав смесей, в состав которых включены среднецепочечные триглицериды (г/100 мл) <1>
———————————
<1> — Также MCT содержатся в ряде смесей для недоношенных детей, полуэлементных смесей на основе глубокого гидролиза белка коровьего молока, элементных смесей на основе аминокислот.
———————————
<2> — Специализированный продукт на основе масла среднецепочечных триглицеридов.
Приложение Г11
ДИЕТА ПРИ НЕДОСТАТОЧНОСТИ TFP/LCHAD (КОНСЕНСУС ЭКСПЕРТОВ)
———————————
<a> Рекомендуемая доля LCT в рационе здоровых младенцев составляет 40 — 45% от общей калорийности, в рационе детей школьного возраста — 30 — 35% (рекомендации D-A-CH)
<b> На практике потребление LCT превышает рекомендованные значения по причине ограниченного выбора продуктов питания; как правило, лишь 15% от общей калорийности обеспечивается MCT, а другие 15% — LCT (включая 3 — 4% незаменимых жирных кислот).
Приложение Г12
ДИЕТА ПРИ НЕДОСТАТОЧНОСТИ VLCAD (КОНСЕНСУС ЭКСПЕРТОВ)
Вариант VLCADD | Возраст | Жиры | Углеводы | Калории |
Бессимптомная (и CK, AST и ALT в пределах нормы) | 0 — 4 месяцев | Половина грудное молоко/детская смесь, половина — специальная смесь с низким содержанием жиров — Monogen, SHS количество на 100 мл: LCT 0,21 г, MCT 1,89 г или | Без специальных добавок (только при клинических показаниях) | Без специальных добавок (рекомендации D-A-CH) |
после 4 месяцев (введение в рацион твердой пищи) | Снижение содержания жиров до 30 — 40% от общей калорийности <a> (рекомендация D-A-CH); 10 — 15% жиров за счет MCT | |||
Симптоматичная | 0 — 4 месяцев | Исключение грудного молока или детских смесей, 100% специальная смесь с низким содержанием жиров) — Monogen, SHS Количество на 100 мл: LCT: 0,21 г, MCT: 1,89 г незаменимые жирные кислоты (3,5 г/день) или — | Без специальных добавок (только при клинических показаниях) | Без специальных добавок (рекомендации D-A-CH) |
после 4 месяцев (введение в рацион твердой пищи) | Снижение содержания жиров до 25 — 30 (-40)% от общей калорийности <a>; 20% жиров за счет MCT <b>; 3 — 4% жиров за счет незаменимых жирных кислот (рекомендации D-A-CH) |
———————————
MCT — среднецепочечные триглицериды; LCT — длинноцепочечные триглицериды; D-A-CH — рекомендации объединения нутриционистов Германии/Австрии/Швейцарии.
<a> рекомендованное потребление LCT для здоровых младенцев составляет 40 — 45% от общей калорийности, для детей школьного возраста — 30 — 35%, согласно рекомендациям D-A-CH/
<b> На практике потребление LCT превышает рекомендованные значения по причине ограниченного выбора продуктов питания; как правило, лишь 15% от общей калорийности обеспечивается MCT, а другие 15% — из LCT (включая 3 — 4% незаменимых жирных кислот).
Приложение Г13
ИСТОЧНИКИ НЕЗАМЕНИМЫХ ЖИРНЫХ КИСЛОТ
[105]
Приложение Г14
СОСТАВ СМЕСЕЙ ДЛЯ ДИЕТИЧЕСКОГО ПИТАНИЯ С ПОНИЖЕННЫМ
СОДЕРЖАНИЕМ LCT
Основные характеристики | Специализированные продукты питания | |
Специализированный продукт диетического лечебного питания для детей с рождения и взрослых при дефектах окисления длинноцепочечных жирных кислот, хилотораксе и лимфангиэктазии — сухая смесь «Monogen» («Моноген») (100 г) | Специализированный продукт детского диетического лечебного питания — «Ликвиджен «**** («Liquigen «) (100 мл) | |
Энергетическая ценность | 1859/441 кДж/ккал | |
Белок | 12,8 г | |
Жиры | 12,9 г | |
Насыщенные ЖК | 11,3 г | |
Мононенасыщенные | 0,34 г | |
Полиненасыщенные | 1,3 г | |
Линолевая кислота (LA) | 0,87 г | |
-Линоленовая кислота (ALA) | 0,17 г | |
Докозагексаеновая кислота (DHA) | 0,06 г | |
Арахидоновая кислота (AA) | 0,06 г | |
Среднецепочечные триглицериды (MCT) | 10,1 г 84,4% (г/100 г жирных кислот) | 50 мл |
Длинноцепочечные триглицериды (LCT) | 2,0 г 15,6% (г/100 г жирных кислот) | |
Отношение (n-6): (n-3) жирных кислот | 3,87:1 | |
Углеводы | 68,6 г | |
Сахара | 11,8 г | |
Вода | 50 мл | |
Осмоляльность | 240 мОсм/кг H2O | |
Приложение Г15
ПРИМЕР РАСЧЕТА ДИЕТЫ У ПАЦИЕНТОВ С НАРУШЕНИЯМИ
ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
Пример N 1 Диагноз: недостаточность длинноцепочечной гидроксил-КоA дегидрогеназы (LCHAD)
Возраст 12 дней (период постановки диагноза)
Масса — 3,2 кг.
1.1 Специализированное питание «Моноген»
1. Отмена грудного вскармливания
2. Переход полностью на специализированную смесь, учитывая, что в данной смеси изначально правильное соотношение жиров, количество смеси рассчитывается только исходя из потребности в энергии.
Расчет диеты:
А) Потребность ребенка в энергии в возрасте в 12 дней составляет 113 ккал/кг/сут.
Рекомендуемая калорийность питания в сутки — 3,2 x 113 = 361,6 ккал
В 100 мл специализированного питания «Моноген» содержится 74,6 ккал,
74,6 ккал — 100 мл
361 ккал — x
X = 361 x 100/74,6 = 484 мл специализированного питания «Моноген» в сутки
Б) Количество кормлений 8 раз в сутки (интервал 3 часа), рассчитываем объем разовой порции:
484 мл / 8 = 60,5 мл
время | Смесь «Моноген» мл. | Общ. Жиры | MCT | ПНЖК | ккал |
6:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
9:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
12:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
15:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
18:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
21:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
24:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
03:00 | 60,0 | 1,32 | 1,08 | 0,13 | 44,76 |
Всего (г) | 10,56 | 8,64 | 1,04 | 358 | |
Всего % | 26,5 | 21,7 | 2,61 |
Пример N 2 Диагноз: недостаточность очень длинноцепочечной ацил-КоA дегидрогеназы (VLCAD) с ранней манифестацией
Возраст 12 мес.
Масса — 12 кг.
Расчет диеты исходя из потребности в энергии, а также соотношения жиров:
А) Рассчитываем потребность в энергии:
В возрасте в 12 мес. Потребность в энергии составляет 80 ккал/кг/сут
Рекомендуемая калорийность питания в сутки — 12 x 80 = 960 ккал
Б) Рассчитываем жиры:
25% — 30% энергии должны обеспечивать жиры, из них 20 — 25%% MCT, рассчитываем необходимое количество в граммах.
Количество общего жира:
25% от 960 ккал — 240 ккал/сут
9 ккал — 1 г жира
240 — x
X = 240 / 9 = 27 г общ. жира
Количество MCT:
20% от 960 ккал — 192 ккал
8,3 ккал — 1 г MCT
192 — x
X = 192 / 8,3 = 23 г MCT
В) Максимальные интервалы между кормлениями 4 часа днем, 6 часов ночью, что соответствует 6 приемам пищи.
Калорийность разового кормления примерно составляет 960 ккал / 6 = 160 ккал
Количество общего жира в разовом кормление примерно составляет 27 г / 6 = 4,5 г
Количество MCT в разовом кормление примерно составляет 23 г / 6 = 3,8 г
Примерное меню для ребенка 12 мес.
Прием пищи | продукт | объем | белки | Общ. Жиры | MCT | ПНЖК | углеводы | ккал |
5:30 | Специализированная смесь «Моноген» | 33,6 (г) 6 м.л. | 4,3 | 4,3 | 3,6 | 0,45 | 23,2 | 148 |
Вода | 180 мл | 0 | 0 | 0 | 0 | 0 | 0 | |
Liquigen**** | 3 мл | 0 | 1,5 | 1,5 | 0 | 0 | 12,5 | |
9:30 | Каша кукурузная (сухая) | 25 г | 1,8 | 0,3 | 0 | 0 | 21 | 93,2 |
Молоко 0,5% 30 мл 170 мл воды | 200 мл | 0,8 | 0,15 | 0 | 0 | 1,5 | 10,5 | |
Liquigen | 8 мл | 0 | 4 | 4 | 0 | 0 | 33,2 | |
Пюре из груши | 50 г | 0,2 | 0 | 0 | 0 | 5,5 | 22,8 | |
Масло грецкого ореха | 1 мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
13:30 | Специализированная смесь «Моноген» | 33,6 (г) 6 м.л. | 4,3 | 4,3 | 3,6 | 0,45 | 23,2 | 148 |
Вода | 180 мл | 0 | 0 | 0 | 0 | 0 | 0 | |
Liquigen**** | 3 мл | 0 | 1,5 | 1,5 | 0 | 0 | 12,5 | |
16:30 | картофель | 100 г | 2,0 | 0 | 0 | 0 | 16 | 72,9 |
морковь | 30 г | 0,3 | 0 | 0 | 0 | 1,7 | 8 | |
кабачки | 30 г | 1 | 0 | 0 | 0 | 0,7 | 6,8 | |
Цветная капуста | 30 г | 0,4 | 0 | 0 | 0 | 1,3 | 7,7 | |
Филе индейки | 30 г | 5,8 | 0,5 | 0 | 0 | 0 | 27,7 | |
Liquigen | 8 мл | 0 | 4 | 4 | 0 | 0 | 33,2 | |
Масло грецкого ореха | 1 мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
20:00 | Творог обезжиренный 0,2% | 100 г | 16 | 0,1 | 0 | 0 | 1,8 | 72,1 |
Яблочное пюре | 100 г | 0,2 | 0 | 11,3 | 46 | |||
Liquigen**** | 8 мл | 0 | 4 | 4 | 0 | 0 | 33,2 | |
Масло грецкого ореха | 1 мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
24:00 | Специализированная смесь «Моноген» | 33,6 (г) 6 м.л. | 4,3 | 4,3 | 3,6 | 0,45 | 23,2 | 148 |
Вода | 180 мл | 0 | 0 | 0 | 0 | 0 | 0 | |
Liquigen**** | 3 мл | 0 | 1,5 | 1,5 | 0 | 0 | 12,5 | |
итог | всего | 41,4 | 33,45 | 27,3 | 3,45 | 130,5 | 975,8 | |
На кг веса | 3,45 | 2,78 | 2,27 | 0,28 | 10,8 | 81,3 | ||
% | 16,9 | 30,8 | 25,1 | 3,2 | 53,4 | |||
Пример N 3 Диагноз: недостаточность очень длинноцепочечной ацил-КоA дегидрогеназы (VLCAD), бессимптомная форма, ребенок находится на грудном вскармливании.
Возраст 2 мес.
Масса — 4,8 кг.
1. Ограничение грудного вскармливание до 50% от общего объема кормлений.
2. Прием специализированной смеси с низким содержанием жиров «Моноген»
Количество смеси рассчитывается исходя из потребности в энергии.
3.1 Специализированная смесь «Моноген»
Расчет диеты:
А) Потребность ребенка в энергии в возрасте в 2 мес. составляет 104 ккал/кг/сут.
Рекомендуемая калорийность питания в сутки — 4,8 x 104 = 500 ккал: 250 ккал за счет специализированного питания/ккал за счет грудного молока.
Количество кормлений 8 раз в сутки (интервал 3 часа)
Б) Рассчитываем специализированное питание — цель покрыть энергию 250 ккал:
74,6 ккал — в 100 мл специализированного питания «Моноген»
250 — x
X = 250 x 100 / 74,6 = 335 мл в сутки
Б) Рассчитываем грудное молоко:
70 ккал — в 100 мл грудного молока
250 — x
X = 250 x 100 / 74,6 = 360 мл в сутки
Б) Количество кормлений 8 раз в сутки (интервал 3 часа), рассчитываем объем разовой порции: 484 мл / 8 = 60,5 мл
время | Смесь «Моноген» мл. | Грудное молоко | Общ. Жиры | MCT | ПНЖК | ккал |
6:00 | 42 | 45 | 2,7 | 0,75 | 0,4 | 62,8 |
9:00 | 42 | 45 | 2,7 | 0,75 | 0,4 | 62,8 |
12:00 | 42 | 45 | 2,7 | 0,75 | 0,4 | 62,8 |
15:00 | 42 | 45 | 2,7 | 0,75 | 0,4 | 62,8 |
18:00 | 42 | 45 | 2,7 | 0,75 | 0,4 | 62,8 |
21:00 | 42 | 45 | 2,7 | 0,75 | 0,4 | 62,8 |
24:00 | 42 | 45 | 2,7 | 0,75 | 0,4 | 62,8 |
03:00 | 42 | 45 | 2,7 | 0,75 | 0,4 | 62,8 |
Всего (г) | 336 | 360 | 21,6 | 6 | 3,2 | 502 |
Всего % | 38 | 10 | 5,7 |
Пример N 4 Диагноз: недостаточность очень длинноцепочечной ацил-КоA дегидрогеназы (VLCAD), бессимптомная форма, ребенок находится на грудном вскармливании.
Возраст 6 мес.
Масса — 8 кг
Расчет диеты исходя из потребности в энергии, а также соотношения жиров:
А) Рассчитываем потребность в энергии:
В возрасте в 6 мес. Потребность в энергии составляет 82 ккал/кг/сут
Рекомендуемая калорийность питания в сутки — 8 x 82 = 656 ккал
Б) Рассчитываем жиры:
35% энергии должны обеспечивать жиры, из них 12% MCT, рассчитываем необходимое количество в граммах.
Количество общего жира:
35% от 656 ккал — 230 ккал/сут
9 ккал — 1 г жира
230 — x
X = 230 / 9 = 25,5 г общ. жира
Количество MCT:
12% от 656 ккал — 78,2 ккал
8,3 ккал — 1 г MCT
78,2 — x
X = 78,2 / 8,3 = 9,4 г MCT
В) Максимальные интервалы между кормлениями 4 часа днем, что соответствует 6 приемам пищи.
Калорийность разового кормления примерно составляет 656 ккал / 6 = 109 ккал
Количество общего жира в разовом кормление примерно составляет 25,5 г / 6 = 4,25 г
Количество MCT в разовом кормлении примерно составляет 9,4 / 6 = 1,57 г
Примерное меню для ребенка 6 месяцев
Прием пищи | продукт | объем | белки | Общ. Жиры | MCT | ПНЖК | углеводы | ккал |
6:00 | Специализированная смесь «Моноген» | 14 г (2,5 мл) | 1,8 | 1,8 | 1,5 | 0,18 | 9,6 | 61,7 |
Вода | 75 мл | 0 | 0 | 0 | 0 | 0 | 0 | |
Грудное молоко | 70 мл | 0,7 | 2,8 | 0 | 0,7 | 4,9 | 49 | |
10:00 | Каша кукурузная (сухая) | 20 г | 1,4 | 0,2 | 0 | 0 | 16,8 | 74,6 |
Грудное молоко 30мл 130 мл воды | 160 мл | 0,4 | 1,2 | 0 | 0,3 | 2,1 | 21 | |
Liquigen**** | 4 мл | 0 | 2 | 2 | 0 | 0 | 16,6 | |
14:00 | Специализированная смесь «Моноген» | 14 г (2,5 мл) | 1,8 | 1,8 | 1,5 | 0,18 | 9,6 | 61,7 |
Вода | 75 мл | 0 | 0 | 0 | 0 | 0 | 0 | |
Грудное молоко | 70 мл | 0,7 | 2,8 | 0 | 0,7 | 4,9 | 49 | |
18:00 | Каша рисовая (сухая) | 20 г | 1,6 | 0,3 | 0 | 0 | 15 | 69,1 |
Вода 150 мл | 30 г | 0 | 0 | 0 | 0 | 0 | 0 | |
брокколи | 50 г | 0,5 | 0 | 0 | 0 | 2 | 10 | |
Liquigen | 6 мл | 0 | 3 | 3 | 0 | 0 | 24,9 | |
22:00 | Специализированная смесь «Моноген» | 14 г (2,5 мл) | 1,8 | 1,8 | 1,5 | 0,18 | 9,6 | 61,7 |
Вода | 75 мл | 0 | 0 | 0 | 0 | 0 | 0 | |
Грудное молоко | 70 мл | 0,7 | 2,8 | 0 | 0,7 | 4,9 | 49 | |
02:00 | Специализированная смесь «Моноген» | 14 г (2,5 мл) | 1,8 | 1,8 | 1,5 | 0,18 | 9,6 | 61,7 |
Вода | 75 мл | 0 | 0 | 0 | 0 | 0 | 0 | |
Грудное молоко | 70 мл | 0,7 | 2,8 | 0 | 0,7 | 4,9 | 49 | |
итог | всего | 13,9 | 25,1 | 11 | 3,8 | 93,9 | 659 | |
на кг веса | 1,7 | 3,13 | 1,37 | 0,47 | 11,7 | |||
% | 8,4 | 34,2 | 13,8 | 5,2 | 57 | |||
Пример 5 Диагноз: недостаточность длинноцепочечной гидроксил-КоA дегидрогеназы (LCHAD)
Возраст 4 года
Масса — 16 кг.
Расчет диеты исходя из потребности в энергии, а также соотношения жиров:
А) Рассчитываем потребность в энергии:
В возрасте 4 лет потребность в энергии составляет 79,7 ккал/кг/сут
Рекомендуемая калорийность питания в сутки — 16 x 79,7 = 1275 ккал
Б) Рассчитываем жиры:
25% — 30% энергии должны обеспечивать жиры, из них 20% — 25% MCT, рассчитываем необходимое количество в граммах.
Количество общего жира:
30% от 1275 ккал — 382,5 ккал/сут
9 ккал — 1 г жира
382,5 — x
X = 382,5 / 9 = 42,5 г общ. жира
Количество MCT:
25% от 1275 ккал — 318,7 ккал
8,3 ккал — 1 г MCT
318,7 — x
X = 318,7 / 8,3 = 38 г MCT
В) Максимальные интервалы между кормлениями 4 часа днем, до 10 часов ночью, что соответствует 5 приемам пищи.
Калорийность разового кормления примерно составляет 1275 ккал / 5 = 255 ккал
Количество общего жира в разовом кормлении примерно составляет 35 г / 5 = 7 г
Количество MCT в разовом кормлении примерно составляет 30 г / 5 = 6 г
Прием пищи | продукт | объем | белки | Общ. Жиры | MCT | ПНЖК | углеводы | ккал |
7:00 | Каша кукурузная | 35 г | 2,5 | 0,3 | 0 | 0 | 29,4 | 130 |
Молоко 0,5% 50 мл 200 мл воды | 250 мл | 1,4 | 0,25 | 0 | 0 | 2,5 | 17,9 | |
Liquigen**** | 12 мл | 0 | 6 | 6 | 0 | 0 | 50 | |
Фрукты (груша) | 100 г | 0,4 | 0 | 0 | 0 | 11 | 47,4 | |
мед | 10 г | 0 | 0 | 0 | 0 | 8,2 | 32,8 | |
Масло грецкого ореха | 1 мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
11:00 | картофель | 150 г | 3 | 0 | 0 | 0 | 24 | 109 |
брокколи | 30 г | 1 | 0 | 0 | 0 | 0,8 | 7,2 | |
Цветная капуста | 30 г | 0,8 | 0 | 0 | 0 | 0,7 | 6 | |
морковь | 30 г | 0,3 | 0 | 0 | 0 | 1,7 | 8 | |
Индейка (вареная) грудка | 50 г | 9,6 | 0,6 | 0 | 0 | 0,2 | 44,6 | |
Масло грецкого ореха | 1 мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
Liquigen**** | 14 мл | 0 | 8 | 8 | 0 | 0 | 66,4 | |
15:00 | Творог обезжиренный 0,2% | 180 г | 28 | 0,4 | 0 | 0 | 3,2 | 127,6 |
банан | 50 г | 0,6 | 0 | 0 | 0 | 10 | 43,3 | |
груша | 50 г | 0,2 | 0 | 0 | 0 | 5,5 | 23,7 | |
Liquigen**** | 14 мл | 0 | 8 | 8 | 0 | 0 | 66,4 | |
Масло грецкого ореха | 1 мл | 0 | 1 | 0 | 0,7 | 0 | 9 | |
19:00 | картофель | 200 | 4 | 0 | 0 | 0 | 32 | 145,8 |
Томат | 50 | 0,5 | 0 | 0 | 0 | 1,3 | 7,2 | |
огурец | 50 | 0,3 | 0 | 0 | 0 | 0,9 | 4,8 | |
Филе индейки | 30 г | 5,8 | 0,4 | 0 | 0 | 0,1 | 27,2 | |
Liquigen**** | 14 мл | 0 | 8 | 8 | 0 | 0 | 66,4 | |
Масло грецкого ореха | 1 мл | 0 | 0 | 0 | 0,7 | 0 | 9 | |
22:30 | Каша рисовая | 35 г | 2,8 | 0,4 | 0 | 0 | 28 | 126,8 |
Молоко 0,5% 50 мл 200 мл воды | 250 мл | 1,4 | 0,25 | 0 | 0 | 2,5 | 17,9 | |
тыква | 50 г | 0,5 | 0 | 0 | 0 | 2,3 | 12,1 | |
мед | 10 г | 0 | 0 | 0 | 0 | 8,2 | 32,8 | |
Liquigen**** | 14 мл | 0 | 8 | 8 | 0 | 0 | 66,4 | |
итог | всего | 63,1 | 43,6 | 38 | 2,8 | 172,2 | 1324 | |
На кг веса | 3,9 | 2,7 | 2,37 | 0,17 | 10,7 | 79,1 | ||
% | 19 | 29,6 | 24 | 1,9 | 52 | |||
Приложение Г16
ЧАСТОТА ПРОВЕДЕНИЯ ОБСЛЕДОВАНИЯ У ПАЦИЕНТОВ С НАРУШЕНИЯМИ
ОКИСЛЕНИЯ ЖИРНЫХ КИСЛОТ
Исследования | При постановке диагноза | Каждые 3 мес | Каждые 6 мес | Каждый год | По показаниям |
Лабораторные исследования | |||||
Определение ацилкарнитинов в крови | X | каждые 1 — 2 месяца пациентам до года | каждые 3 — 6 месяцев у детей младше 6 лет | каждые 6 — 12 месяцев пациентам старше 6 лет | В период и при подозрении на метаболический криз |
Определение содержания органических кислот в моче | X | ||||
Молекулярно-генетическое исследование | X | ||||
Общий (клинический) анализ крови развернутый | X | X | X | ||
Анализ крови биохимический общетерапевтический | X | X | В период и при подозрении на метаболический криз | ||
Анализ кислотно-щелочного состояния крови (Исследование уровня буферных веществ в крови, Исследование уровня водородных ионов (pH) крови), глюкозы, электролитов (калий, натрий, кальций ионизированный, хлор) | В период и при подозрении на метаболический криз | ||||
Общий (клинический) анализ мочи, определение кетоновых тел | X | X | X | X | |
NT-proBNP | X | ||||
Инструментальные исследования | |||||
Электрокардиограмма | X | X | |||
Эхокардиография | X | X | |||
УЗИ органов брюшной полости; объем селезенки, печени; УЗИ почек | X | X | X (при наличии патологии чаще) | ||
Осмотр глазного дна (офтальмоскопия) | X | X | X (при наличии патологии чаще) | ||
Электроэнцефалография (ЭЭГ) | X | X (при наличии эпилептических приступов чаще) | |||
Консультации специалистов | |||||
Консультация врача-генетика | X | X | |||
Консультация врача-педиатра/терапевта | X | X | |||
Консультация врача-невролога | X | X | |||
Консультация врача-диетолога | X | ||||
Консультация врача-гастроэнтеролога | X | ||||
Консультация врача-офтальмолога | X | X | |||
Консультация врача-кардиолога, врача-детского кардиолога | X | X | |||
Консультация врача-нефролога | X | ||||
Консультация врача-эндокринолога | X | ||||
Консультация/консультации медицинского психолога | X | X | |||
Консультация врача-физиотерапевта и врача по лечебной физкультуре | X | ||||
Приложение Г17
ПРИМЕР «ЭКСТРЕННОЙ ПАМЯТКИ»
Экстренная памятка
Внимание! Пациент угрожаем по развитию острого метаболического криза, связанного с наследственным заболеванием.
1. Показания к госпитализации:
— многократная рвота, диарея;
— гипогликемия;
— интеркуррентные инфекции с высокой лихорадкой;
— обезвоживание;
— выраженная слабость или чрезмерная сонливость;
— мышечные боли, слабость или красновато-коричневый цвет мочи
2. План действия:
2.1. Суточная калорийность питания 110 — 120% от обычного рациона.
2.2. При нахождении ребенка дома, увеличить потребление углеводов для восполнения калорийности за счет раствора декстрозы или мальтодекстрина (при их отсутствии — сладкий чай, сок, компот)
Соответствие между возрастом ребенка и дозой декстрозы для энтерального применения при подозрении на метаболический криз.
2.3. При госпитализации в стационар, начать непрерывную инфузию р-ром 10% декстрозы** из расчета: новорожденные и дети до 3-х лет 10 — 12 мг/кг/мин, от 3 до 10 лет 8 — 10 мг/кг/мин, старше 10 лет 5 — 8 мг/кг/мин
2.4. Контроль анализов:
— КЩС, электролиты, аммоний
— Анализ крови биохимический общетерапевтический (КФК, АЛТ, АСТ, глюкоза, билирубин общ. прямой, мочевина, креатинин, ГГТ, ЩФ, общий белок, альбумин) спектр ацилкарнитинов — спектр аминокислот и ацилкарнитинов методом тандемной масс-спектрометр
3. Ведение пациента после неотложной ситуации:
— При невозможности осуществлять, поддерживать пероральное кормление, установить назогастральный зонд
— Продолжить инфузию растворами декстрозы до стабилизации состояния, под контролем глюкозы крови и КЩС
4. Противопоказано:
— Внутривенное введение жировых растворов
— Внутривенное введение карнитина за исключением OCTN2 карнитина
— Голодание при отмене в/в инфузий
Приложение Г18
ПЕРЕЧЕНЬ ОСНОВНЫХ ЛАБОРАТОРНЫХ ИССЛЕДОВАНИЙ, КОТОРЫЕ МОГУТ
ПОМОЧЬ В ОКАЗАНИИ МЕДИЦИНСКОЙ ПОМОЩИ ПРИ СОСТОЯНИИ
ОСТРОЙ МЕТАБОЛИЧЕСКОЙ ДЕКОМПЕНСАЦИИ
———————————
<*> При отправке лабораторных анализов необходимо руководствоваться клинической картиной пациента. Это не исчерпывающий список, и некоторые из перечисленных здесь тестов могут не указывается.
МНО — международное нормализованное отношение, КК-МВ — МВ-фракция креатинкиназы; МНП — мозговой натрийуретический пептид; Эхо-КГ — Эхокардиография; ОАК — общий анализ крови;
Приложение Г19
РАСШИФРОВКА ПРИМЕЧАНИЙ
…** — препарат входит в перечень жизненно необходимых и важнейших лекарственных препаратов (Распоряжение Правительства РФ от 12.10.2022 N 2406-р «Об утверждении перечня жизненно необходимых и важнейших лекарственных препаратов на 2020 год, а также перечней лекарственных препаратов для медицинского применения и минимального ассортимента лекарственных препаратов, необходимых для оказания медицинской помощи», Распоряжение Правительства РФ от 23 ноября 2020 г. N 3073-р
# — применение off-label — вне зарегистрированных в инструкции лекарственного средства показаний осуществляется по решению врачебной комиссии, с разрешения Локального этического комитета медицинской организации (при наличии), с условием подписанного информированного согласия родителей (законного представителя) и пациента в возрасте старше 15 лет;
…**** — специализированный продукт лечебного питания, включенный в перечень специализированных продуктов лечебного питания для детей-инвалидов (Распоряжение Правительства РФ от 7 декабря 2020 г. N 3242-р Об утверждении перечня специализированных продуктов лечебного питания для детей-инвалидов на 2021 г.)
Переработка молока — «окисление липидов и порча молочных продуктов»
Окисление молочного жира и фосфолииидов молока вызывается ферментами, но чаще происходит химическим путем — под действием кислорода воздуха и света. Как правило, окисление липидов снижает биологическую ценность молока и молочных продуктов и часто вызывает их порчу.
Перекисное окисление жира. Под окислением жира следует понимать его глубокий распад с образованием перекисей (пероксидов), альдегидов, кетонов, оксикислот и других соединений, которые очень часто приводят к появлению в молочных продуктах нежелательных привкусов и запахов.
Перекисное окисление происходит в результате взаимодействия жира с молекулярным кислородом. Окислению подвергается в первую очередь свободный жир, не защищенный оболочкой, а из жирных кислот преимущественно окисляются ненасыщенные. Окисление свободных и свйзанных жирных кислот молекулярным кислородом проходит через цепные реакции с образованием промежуточных продуктов пе- рекисного типа. Существенную роль в начальной стадии перекисного окисления играют свободные радикалы — радикалы, один из атомов которых имеет свободную валентность. Теория свободнорадикальных ценных реакций окисления липидов разработана акад. Н. Н. Семеновым на основе теории перекисного окисления Баха—Энглера.
Согласно современным представлениям образование свободных радикалов, приводящих к зарождению {инициированию) цепи окисления, происходит при отрыве атома водорода от реагирующей молекулы вещества (жир, жирная кислота): RH R’ Н. Инициатором цепных реакций могут быть металлы, кислород, ферменты, свет, различные типы излучения (ультрафиолетовое, радиация и др.) и Т. II.
Далее активный радикал R вступает в реакцию с молекулярным кислородом, образуя пероксидный радикал R’ О, ROO’. Перок- сидный радикал, реагируя с новой молекулой окисляемого вещества, дает гидроперекись (гидропероксид) и новый свободный радикал ROO’ RH – г ROOH R*.
Образовавшийся свободный радикал R» вновь реагирует с кислородом, то есть происходит продолжение (развитие) цепи*:
о, кн R»— ^ROO’ jfc – ROOH R’ и т. д.
Молекулы гидропероксидов в свою очередь распадаются с обра» зованием новых свободных радикалов: ROOH R0′ ‘ОН. Когда концентрация гидропероксидов повышается, происходит их распад с образованием еще большего числа радикалов: 2ROOH —» ROO’ R0″ HjO. Эти радикалы способствуют зарождению новых цепей окисления, вызывая тем самым самоускорение процесса окисления жира.
Таким образом, окисление жиров молекулярным кислородом можно представить схематически следующим образом (по Уильямсу):
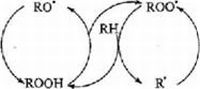
* Свободные радикалы могут таюке взаимодействовать с другими радикалами, образуя стабильные продукты я вшивая обрыв цепи окисления: R’ R’ – t KR, R’ ROO’ — ROOR. Анало гично обрывают цепь оклелення антиоксиланты.
Скорость окисления жира в первую очередь зависит от состава жирных кислот триацилглииеринов (причем свободные жирные кислоты окисляются быстрее связанных). Насыщенные жирные кислоты окисляются медленнее ненасыщенных, а полиненасыщенные — быстрее мононенасыщенных, что объясняется различной скоростью образования ими свободных радикалов.
Окисление ненасыщенной жирной кислоты можно представить следующим образом. Вначале кислота под влиянием свста или другого инициатора образуется свободный радикал
R-CH2-CH=CH-R2—СООН R|-CH-CH=CH-RJ-COOH.
Свободный радикал взаимодействует с кислородом, образуя пе – роксидный радикал
R^Ii—СН=СН—R^COOH Ог———— — R, СН-СН=СН-R^COOH.
У-6
Пероксидный радикал отщепляет атом водорода от другой молекулы ненасыщенной жирной кислоты, обра^я гидропероксид и новый свободный радикал, который продолжает цепную реакцию
RjCH-CH-CH-RjCOOH RнCH-CH=CH-RMCOOH———————
O-У
—— ^R1CII-CH=CH-R^COOH R3CH-CH=CH-R4COOH.
О-он
Следовательно, на первой стадии окисления образуются различные пероксиды и гидропероксиды, являющиеся неустойчивыми и высокоактивными соединениями. Первичные продукты окисления существенно не влияют на органолептические свойства продуктов. После их накопления в жире начинают протекать разнообразные реакции, в результате которых образуются вторичные продукты окисления, часто обладающие неприятным вкусом и запахом, — альдегиды, кетоны, моно – и дикарбоновые кислоты, эпоксиды, окси – сосдинения и т. д.
Одним из возможных механизмов, приводящих к образованию наиболее характерных продуктов окисления — альдегидов, является распад гидропероксидов иди циклических пероксидов кислот по схеме
Яг-СН2-СН-СН-СН2-К2–СООН—»-
I I
О—О р
— л,—СН2-С С»СН2-^-СООН / н н
Альдегид
В результате распада образуются два альдегида, молекулярные массы которых меньше массы исходной кислоты. Образование насыщенных и ненасыщенных кетонов (и кетокислот) можно представить как результат дегидратации гидроксидов
| ~н20 1 1 II *
Он о
Кеток (или кето кислота.)
Образование оксисоединений (дигидроксисоединений) может происходить при распаде циклических пероксидов
2Н20
Я-СН2-СН-СН-СНг^2—- к-сн2-сн-сн-сн2-к2 Н202
О—О ОН ОН
Дигидроксисоединение
Таким образом, при окислении олеиновой и полиненасыщенных кислот могут образоваться низкомолекулярные насыщенные альдегиды — пентаналь, гексаналь, гептаналь, октаналь, нонаналь, малоновый диальдегид и другие мононенасыщенные и диненасы- щенные альдегиды — пентен-2-аль, октен-2-аль, гептадиен-2,4-аль, декадиен-2,4-аль, а также насыщенные и ненасыщенные кетоны (С5,мС15). Многие из перечисленных альдегидов и кетонов обладают неприятным вкусом и в различных комбинациях могут придавать молочным продуктом посторонние привкусы. Так, рыбный привкус вызывают насыщенные и ненасыщенные альдегиды (С5…СП), главным образом гексаналь, гептанальи гептадиен-2,4-аль; прогорклый — гепганаль и нонаналь; салистый — пентаналь, гексаналь, пенген-2-аль {и дигидроксистеариновая кислота). Ненасыщенный кетон октен-1-он-З (виниламилкетон) является виновником металлического привкуса молочных продуктов, а кетон пептен-1-он-З — олеистого привкуса.
Состав образующихся продуктов и скорость окисления жира молекулярным кислородом зависят от целого ряда факторов: химического состава жира, температуры хранения, влажности и т. д. На процесс окисления жира влияют некоторые химические вещества, которые либо ускоряют его (прооксиданты), либо замедляют (антиокси – данты).
Скорость окисления жира увеличивается при повышении температуры, влажности, доступе в реакционную среду кислорода воздуха, света и т. д. Сильно ускоряют окисление жира металлы переменной валентности (Cu, Fe, Со, Мп и др.), которые относятся к основным прооксидантам. Их ускоряющее действие может заключаться, во-первых, в инициировании цепей окисления; во-вторых, возможный механизм ускорения окисления жиров может заключаться в катализи- ровании процесса распада гидропероксидов, продукты которого дают начало новым цепям окисления:
ROOH Fe2 ——– >~ RO’ ОН» FeJ%
RO’ RH——— ROH R’———— Цепное окисление
Металлы проявляют прооксидантное действие только при малом содержании, в больших концентрациях они могут обрывать цепи окисления и замедлять окисление жира.
Задержку’ окислительной порчи продуктов вызывают так называемые антиоксиданты, или антиоки&штели. Действие ан-гиоксидан – тов заключается во взаимодействии со свободными радикалами, ведущими цепи окисления. В результате этого происходит обрыв цепей окисления и на какой-то период времени задерживается процесс самоокисления жира:
ROO’ АН——– ROOH А’
Или R – АН——- RH А»,
Где А — антиоксидант.
Из приведенных реакций видно, что активные свободные радикалы ROO» и R» заменяются на малоактивные радикалы аити – оксиданта А». Последние подвергаются димернзации или вступают в реакцию с другими радикалами, образуя неактивные продукты:
А’ А—- A’ R————— – RA
Или А’ ROO*———- ROOA
Известно большое количество соединений, обладающих сильными антиоксидантными свойствами. Одни из них содержатся в пищевых продуктах (естественные, или природные антиоксидан – ты), другие получают искусственно и вносят в продукты (синтетические антиоксиданты). Среди естественных антиоксидантов наиболее активными являются токоферолы, содержащие подвижный атом водорода, который может взаимодействовать со свободными радикалами R’. При потере атома водорода токоферолы образуют малоактивные радикалы, прерывающие цепь окисления:
К менее активным природным антиокислителям относятся аскорбиновая кислота, 8Н-соединения, р-каротин, лецитин и др. Кроме того, некоторые соединения могут усиливать действие естественных антиоксидантов. К таким соединениям, называемым си – нергистами, относится лимонная кислота, а также аскорбиновая и винная кислоты, лактаты калия и натрия, фосфолипиды. Их синер – гетическое действие заключается в восстановлении окисленных форм антиоксидантов или в связывании ионов тяжелых металлов в неактивные комплексы.
Из синтетических антиоксидантов наибольшую активность имеют соединения фенольноготипа — эфиры галловой кислоты (пропил галл ат, октилгаллат, додецилгаллат и др.), бутилгидрок – сианизол (БОА), бутилгидрокситолуол (БОТ, ионол), кверцетин и др.
До недавнего времени синтетические антиокислители в РФ использовали только для защиты кулинарных, животных (свиного, говяжьего и др.) и рыбьих жиров, сухого молока. В настоящее время появился отечественный антиоксидант тонарол (4-метил-2,6-ди’фетбугил-фе – нол, аналогионода), который разрешено Госкомсанэпиднадзором вносить в сливочное масло для повышения ею хранимоспособности.
Окисление фосфолипидов. Фосфолипиды относительно легко окисляются кислородом воздуха, особенно при наличии в молоке и молочных продуктах солей тяжелых металлов. В первую очередь окисляются фосфолипиды, находящиеся в плазме молока, затем — фосфолипиды оболочек шариков жира. Активное окисление фосфолипидов обусловлено высоким содержанием в их молекулах полиненасыщенных жирных кислот — линолевой, линоленоаой и арахидоно – вой. Данные жирные кислоты окисляются кислородом воздуха с образованием пероксидов и карбонильных соединений, вызывающих ухудшение органолептических свойств молочных продуктов.
Как известно, фосфолипиды являются важными структурными компонентами оболочек шариков жира, поэтому их окисление будет способствовать дестабилизации жировой фазы и лучшей атакуемо – сти жира кислородом воздуха.
Окислительная порча молочных продуктов. Возникновение пороков вкуса и запаха вследствие развития окислительных процессов характерно для молока (сырого и пастеризованного), сливочного масла, сухих молочных продуктов. В других продуктах (сгущенное молоко с сахаром, сгущенное стерилизованное молоко, некоторые виды сыров, сметана, творог и др.) интенсивность образования данных пороков невысокая и возрастает лишь в результате длительного хранения, особенно при воздействии кислорода и света.
Окисление липидов молока. В процессе длительного хранения молока при низких температурах, а также под воздействием светового излучения с длиной волны менее 500 нм в продукте возникают окисленные привкусы — «картонный» и «солнечный», которые иногда сопровождаются металлическим, рыбным и салистым привкусами. Окисленные привкусы молока обусловливаются образованием альдегидов (этан ал я, пропаналя, метионаля, пентакаля и др.), метилке – тонови спиртов.
Развитие окисленного привкуса в молоке ускоряют дестабилизация жировой фазы, ионы меди, железа, аскорбиновая кислота в малых концентрациях.
Дестабилизированный (свободный) жир содержит больше поли – ненасышенных жирных кислот по сравнению с обычным жиром, поэтому скорость его окисления в 1,5„.3 раза выше. Количество свободного жира в молоке зависит от времени года, степени механического воздействия при хранении и других факторов.
Как известно, металлы являются основными прооксидантами, ускоряющими окисление липидов. Прооксидантными свойствами также обладает аскорбиновая кислота в малых количествах.
Предотвращению появления окисленного привкуса в молоке способствуют снижение степени механического воздействия при хранении, устранение действия света, внесение в молоко аскорбиновой кислоты (в количестве 25…50 мг на 1 кг), р-каротина, низина, пастеризация при высоких температурах и гомогенизация молока.
Окисление сливочного масла. Образование пероксидов, альдегидов, кетонов, оксикислот и других соединений при окислении липидов сливочного масла в процессе выработки и особенно при хранении приводит к снижению его качества, биологической ценности и возникновению пороков вкуса и запаха — салистого, прогорклого, рыбного, металлического и олеистого. Границы перекисных чисел для молочного жира не установлены. Как показывает практика, перекис – ные числа стойкого молочного жира (масла) находятся в пределах от 0,03 до 0,08% I, (или немного выше), 11ри окисленности жира по пробе на содержание малонового диальдегида с 2-тиобарбитуровой кислотой (2-ТБК) образны молочного жира считаются свежими при величине оптической плотности (при длине волны 535 нм), равной ОД 10. ..0,064 ед. на I г жира.
Скорость и направленность процесса окисления, а следовательно, и стойкость сливочного масла при хранении зависят от многих факторов. К ним относятся: химический состав, структура масла, объем и состав плазмы, ее дисперсность, содержание в масле воздуха, металлов, хлорида натрия, молочной кислоты, естественных и синтетических антиоксидантов, вид упаковочных материалов, температура хранения.
Химический состав молочного жира значительно влияет на стойкость масла при хранении, особенно содержание в нем полиненасыщенных жирных кислот (линолевой, линоленовой и арахидоновой). Их содержание зависит от времени года {повышается весной, понижается осенью и зимой). Чаще всего нестойко при длительном хранении масло, выработанное из весеннего молока.
Окислительная порча масла протекает, главным образом, на границе фаз жир-вода, жир-воздух. Следовательно, стойкость масла при всех прочих условиях зависит от степени диспергирования влаги (плазмы) и содержания в нем воздуха. С увеличением степени диспергирования влаги устойчивость масла к процессу окисления снижается. Масло, выработанное методом преобразования высокожирных сливок, характеризуется наиболее тонким распределением влаги. Поэтому в процессе хранения при низких отрицательных температурах (-18°С) оно менее устойчиво, чем масло, полученное сбиванием сливок, характеризующееся более крупными каплями плазмы. Однако оно обладает повышенной устойчивостью при более высоких минусовых (—5*С) и плюсовых температурах, когда имеют место не только химические, но и ферментативные процессы.
Ме таллы, особенно медь, снижают устойчивость масла к окислительной порче. Установлено, что масло при содержании меди 0,4 мг/кг приобретает через 3 мес хранения (при температуре -18″С) рыбный привкус, а при количестве 1,5.,,2 мг/кг быстро снижает свое качество (Л. Н – Ловачевидр,).
К факторам, влияющим на стойкость масла при хранении, относится содержание в нем антиокислителей (антиоксидантов), задерживающих окисление жира. Масло летней выработки, богатое естественными антиокислителями (токоферолами, (^-каротином, 5Н-группами) более стойко при хранении, чем масло, выработанное зимой.
Для повышения стойкости масла при длительном хранении необходимо снижать загрязненность сливок и масла медью и железом, вносить р-каротин и синтетический антиоксиданттонарол в количестве 0,05…0,020% к массе жира, предохранить его от контакта с воздухом, светом, влагой, применять в качестве упаковочных материалов полимерные мешки-вкладыши, алюминиевую кашированную фольгу и другие газо-, свсто – и влагонепроницаемые материалы (Ф. А. Вьшгемирский и др.).
Окисление сухих молочных продуктов. Окисление липидов сухого цельного молока (СЦМ) и других сухих продуктов является одним из видов порчи, которое приводит к ухудшению их органолептических свойств и снижению биологической ценности. Окисленный, салистый и другие привкусы СЦМ могут быть вызваны большим содержанием в продукте воздуха (более 0,1%), свободного жира (выше 9%), солей меди и железа (более 10 мг/кг сухого вещества молока).
Количество воздуха в СЦМ обусловлено размерами частиц, содержание свободного жира — количеством дестабилизированного жира в сырье. Оба показателя зависят от режимов сгущения, распыления и сушки молока, скорости охлаждения продукта после выхода из сушильной башни, вида упаковки, температуры хранения и влажности воздуха и т. д.
Устойчивость сухих продуктов к окислению можно повысить пу – темвнесения антиоксидантов (сдобавлением ихсинергистов или без них) — кверцетина, эфиров галловой кислоты, бутилгидрокситолуо – ла, бутилгидроксианизола и других, а также осуществления гомогенизации сгущенного молока перед сушкой, хранения продуктов в атмосфере азота.