
 • Термодинамика — теплота • Онлайн-конвертеры единиц измерения")
В равных объемах различных газов при одинаковых условиях
(t0С , P) содержится одно и то же число молекул.Законсформулирован итальянским физиком Авогадро в 1811г и имеет ряд следствий.
Следствие 1. В одном моле вещества содержится 6, 02 ∙10 23 молекул.
Значение 6, 02 ∙ 10 23 моль –1 называют числом
Авогадро(NA).
Следствие 2. Один моль любого газа при нормальных условиях (н.у.)
С(273 K), 1,03 . 105 Па) занимает объем 22,4 л/моль.
1 моль О2 (М(О2)=32 г/моль) – содержит 6,02•1023 молекул кислорода, он занимает объем 22,4 л/моль.
1 моль Н2 (М(Н2)=2 г/моль) – содержит 6,02•1023 молекул водорода, он занимает объем 22,4 л/моль.
Задача. Какой объем водорода выделится при травлении 3,25 г Zn соляной кислотой?
Для решения задачи записываем уравнение реакции:
Zn 2 HCl → ZnCl2 H2
65 22,4
где 65 – молярная масса цинка; 22,4 – молярный объем водорода.
При травлении 65 г цинка выделится 22,4 л/моль водорода, а при травлении 32,5 г цинка выделится Х л водорода. Найдем объем выделившегося водорода при н.у.
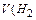 ) = X =
) = X = 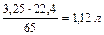
В результате работ немецкого химика Рихтера, английских ученых Дальтона и Волластона (1792 – 1800 г.) были установлены так называемые “соединительные веса” или эквиваленты реагирующих веществ.
Химическим эквивалентом вещества называют такое его количество, которое соединяется полностью с одним моль атомов водорода или замещает такое же количество водорода в его соединениях.
Например, в соединениях НС1, H2О, NH3 эквиваленты хлора, кислорода, азота соответственно равны 1 моль, 1/2 моль, 1/3 моль. За единицу эквивалента принят эквивалент водорода Э (Н) =1 моль атомов). Эквивалент выражают в молях. Кроме понятия эквивалент пользуются понятием молярная масса эквивалента.
Молярная масса эквивалента – это масса одного химического эквивалента вещества МЭ (Н) = 1 г/моль; МЭ (N) = 14∙1/3 = 4,67 г/моль; МЭ (Cl) = 35,45 г/моль; МЭ (S) = 32∙1/2 = 16 г/моль.
Молярная масса эквивалента элемента равна отношению молярной массы элемента (М) к его валентности (В). Например:
S 4O2 , валентность серы равна IV; MЭ (S 4) = M(S)/B = 32/4 = 8 г/моль;
S 6O3, валентность серы равна VI; MЭ (S 6) = M(S)/B = 32/6 = 5,3 г/моль.
Для газообразных веществ удобно пользоваться эквивалентным объемом.
Эквивалентный объем (Vэ) – это объем, занимаемый одним химическим эквивалентом газообразного вещества при нормальных условиях. Эквивалентная масса водорода составляет 1/2 его моля (M(H2)=2 г/моль). Молярный объем водорода равен 22,4 л (н.у.), тогда эквивалентный объем водорода равен 11,2 л/моль. Эквивалентная масса кислорода составляет 1/4 его моля, (M(О2)= 32 г/моль).
Молярный объем кислорода составляет 22,4 л/моль (н.у.), тогда эквивалентный объем кислорода равен 5,6 л/моль.
§
§
Задача 1. Вычислите молярную массу эквивалента металла в оксиде, содержащем 52% металла. Назовите этот металл, если он шестивалентен в данном оксиде. Составьте формулу оксида.
Решение: 100 – 52 = 48% кислорода в оксиде.
По закону эквивалентных отношений записываем выражение:
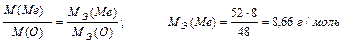
m (Me) = MЭ (Ме)∙В = 8,66∙6 = 52 г/моль
Шестивалентным металлом с атомной массой 52 а.е.м. является хром. Его оксид CrO3.
Задача 2. На восстановление 7,09 г оксида металла требуется 2,24 л водорода (н.у.). Вычислите молярную массу эквивалента металла и его оксида.
Решение.
На основании закона эквивалентных отношений записываем выражение:
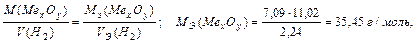
MЭ (Ме xОy) = MЭ (Ме) MЭ (О); (МЭ (О)=8 г/моль), тогда
MЭ (Ме) = 35,45 – 8 = 27,45 г/моль.
Задача 3. Из 3,85 г нитрата металла Me(NO3)x получено 1,60 г его гидроксида Me(OH)x. Вычислите эквивалентную массу металла.
Решение.
На основании закона эквивалентных отношений записываем выражение:
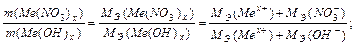

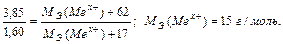
Задача 4. При взаимодействии 3,24 г трехвалентного металла с кислотой выделяется 4,03 л Н2 (н.у.). Вычислите молярную массу эквивалента и молярную массу металла. Назовите металл.
Решение.
Используя закон эквивалентных отношений, запишем выражение:
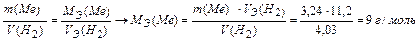
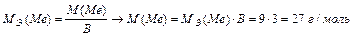
Металл – алюминий.
Знание закона эквивалентных отношений необходимо для:
1. химических расчетов и идентификации веществ.
2. определения молярной концентрации эквивалентов вещества в растворе и использование полученных расчетов в объемном анализе.
§
СН = N = моль/л = 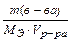
Например,
1 н раствор НС1 → в 1 л раствора содержит 1 молярную массу эквивалента с МЭ(HCl) = 36,5 г/моль.
0,1 н раствор НС1 → в 1 л раствора содержит 0,1 молярной массы эквивалента с МЭ(HCl) = 3,65 г/моль.
0,01 н раствор НС1 → в 1 л раствора содержит 0,01 молярной массы эквивалента с МЭ(HCl) = 0,365 г/моль.
Задача 5. Определить нормальность раствора NaOH, если на титрование 10 мл его расходуется 8,2 мл 0,1 н раствора НС1.
Используя математическое выражение закона эквивалентных
отношений в объемном анализе:N1∙V1 = N2∙V2,записываем выражение:
N(NaOH) ∙V(NaOH)= N(HCl)∙V(HCl)
N(NaOH)= 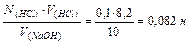
От вет: раствор NaOH 0,082 нормальный, т.е. содержит 0,082молярной массы эквивалента NaOH на один литр раствора (моль/л).
3. Определения жесткости воды:
Жесткость воды показывает количество миллиэквивалентов (мэ/л) растворимых солей кальция и магния, содержащихся в одном литре воды. Ж (Н2О) (мэ/л).
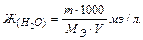
Самостоятельно: виды жесткости воды и способы ее устранения (см. учебник
гл. XIX, § 212, c. 596-599, задачник гл. XI, § 8, с. 232).
Задача 1. Чему равна жесткость воды, в 100 л которой содержится 14,63 г Mg (HCO3)2?
Для решения задачи используем формулу для определения жесткости воды.
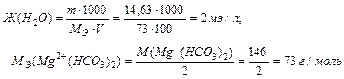
§
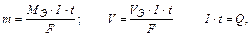
m – масса вещества (г), V – объем вещества(л);
I – сила тока, А;
t– время, с.
F = 96500 Кл/моль (F = 26,8 А∙ч/моль) (Кл = Кулон) ;
Q – количество электричества (А . с; А . ч).
КВАНТОВОМЕХАНИЧЕСКОЕ ПРЕДСТАВЛЕНИЕ О СТРОЕНИИ АТОМА. ПРАВИЛА И ПОРЯДОК ЗАПОЛНЕНИЯ АТОМНЫХ ОРБИТАЛЕЙ.
План:
1. Различные теории о строении атома.
2. Квантовые числа, их физический смысл.
3. Принципы заполнения атомных орбиталей.
Литература:
1. Фролов В.В. Химия. Гл.V, §51-56.
2. Лучинский Г.П. Курс химии. Гл.V, §8-12, гл. VI, §13-18
3. Ахметов Н.С. Общая и неорганическая химия. Раздел V, гл.3,4.
4. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
Научные основы атомно-молекулярного учения были заложены в работах русского ученого М.В.Ломоносова, французских химиков Лавуазье и Пруста, английского химика Дальтона и т.д. Однако до начала двадцатого века, атом считался единым и не делимым. Лишь только серия открытий 1896 – 1898 гг естественной радиоактивности Анри Беккерелем (1896 г — радиоактивность урана), Марией Склодовской-Кюри и Пьером Кюри, позволила изменить представления о неделимости атома.
Атом – это электронейтральная частица, состоящая из положительно заряженного ядра и отрицательно заряженных электронов ( 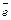 ), вращающихся вокруг него. Ядра атомов имеют сложное строение и состоят из нуклонов (протонов (
), вращающихся вокруг него. Ядра атомов имеют сложное строение и состоят из нуклонов (протонов ( 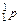 ) и нейтронов (
) и нейтронов ( 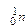 )).
)).
Нуклоны
Атом [Ядро[∑ ∑ ] ∑ē]← Субатомарные частицы
массовое число (А)
Масса ядра меньше суммы масс протонов и нейтронов. Эта разность называется дефектом массы. Он характеризует устойчивость атомных ядер и энергию связи нуклонов в ядре (7.106 эВ), которая в миллионы раз превышает энергию связи атомов в молекуле (5 эВ). Поэтому при химических реакциях ядра атомов не изменяются.
Таблица № 1
| Система СИ | Система атомных единиц | ||||
| Наименование субатомарной частицы | Масса, кг | Заряд, Кл | Масса, а.е.м. | Заряд, а.е.з. | |
| Электрон | 9,109•10-31 | 1,602•10-19 | 0,0005486 | -1 | |
| Нук- лоны | Протон | 1,673•10-27 | 1,602•10-19 | 1,007277 | 1 |
| Нейтрон | 1,673•10-27 | 1,008695 |
Вид атомов с одинаковым зарядом ядра и, следовательно, с одинаковым атомным номером называется химическим элементом.
Каждый химический элемент имеет несколько изотопов.
Атомы с одинаковым зарядом ядра, но разными массовыми числами называют изотопами.
Таблица №2
Кроме изотопов существуют изотоны и изобары.
Изотоны — это атомы с одинаковым числом , но различным
количеством (т.е. с разным зарядом ядра).
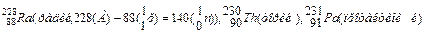 ( =140)
( =140)
Изобары – это атомы с одинаковыми массовыми числами, но разными атомными номерами, например,
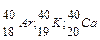
В период открытия первых трех фундаментальных частиц (е; ; ) ,был выдвинут целый ряд моделей строения атома.
1. 1903 г. Модель Д.Томсона, В. Томсона (модель « пудинга с изюмом»), согласно которой в положительно заряженную сферу атома вкраплены электроны:
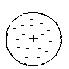 Рис. 1
Рис. 1
2. 1911 г. Э.Резерфорд в результате знаменитых экспериментов по рассеянию золотой фольгой α — частиц установил, что атом:
— имеет достаточно массивное положительно заряженное ядро, имеющее очень малые размеры, которое окружено электронами;
— атом электронейтрален, т.е. положительный заряд ядра численно равен отрицательному суммарному заряду электронов, окружающих это ядро;
— заряд ядра численно равен порядковому номеру элемента в периодической системе Д.И.Менделеева.
Рис.2
Однако, согласно законам классической механики и электродинамики, вращение электрона вокруг ядра должно сопровождаться электромагнитным излучением с непрерывным спектром, что противоречило известным с 1880г. линейчатым спектрам газов и паров элементов. Противоречие разрешил ученик Резерфорда Нильс Бор.
3. 1913 г. Н. Бор разработал планетарную модель атома, которая используется и в наше время. Подобно Резерфорду, Бор представлял себе атом в виде положительно заряженного ядра, окруженного электронами. Однако он предположил, что электроны двигаются вокруг ядра по устойчивым круговым орбитам. Этим орбитам соответствуют различные энергии. Переходя с одной орбиты на другую, электроны могут приобретать либо терять энергию. Н. Бору удалось объяснить и рассчитать теоретически линейчатые спектры испускания атомов водорода, а также серии линий в рентгеновских спектрах элементов.
рис.3
4. 1924 г. Луи де Бройль показал, что элементарная частица, движущаяся с определенной скоростью, может рассматриваться не только как частица, обладающая массой покоя, но и как волна с определенной частотой колебаний (n), удовлетворяя условию равенства энергий:
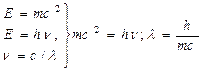
h=6,626 . 10 –34 Дж . с (постоянная Планка);
с=3 . 10 8 м/с (скорость света);
λ=h/mc (взаимосвязь длины волны (λ) и массы частицы (m) обратнопропорциональная).
Результатом работ де Бройля, Дирака, Гейзенберга, Шредингера и других было создание новой физической теории — КВАНТОВОЙ МЕХАНИКИ, которая заявила о корпускулярно-волновом дуализме микрочастиц (например, электрон имеет массу покоя 9,109•10-31 кг., проявляя свойства частицы, а в опытах по дифракции он проявляет свойства волны). В квантовой механике классическое понятие «траектория» заменяется понятием «волновая функция ψ» или «атомная орбиталь (АО)».
АО – это область около ядерного пространства, где электрон может находиться с достаточно высокой степенью вероятности. Термин орбиталь созвучен термину орбита, однако смысл их различен. Орбита — это траектория движения, атомная орбиталь – волновая функция. Если волновая функция (ψ) частицы известна, можно рассчитать вероятность (ψ2) нахождения частицы в различных областях пространства.
В 1925 г. Эрвин Шредингер предложил уравнение, позволяющее математически описывать волновые функции частиц.
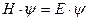 — где Н – Гамильтон
— где Н – Гамильтон 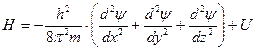
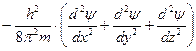 — оператор кинетической энергии,
— оператор кинетической энергии,
U — оператор потенциальной энергии.
Это дифференциальное линейное уравнение второго порядка в частных производных имеет бесчисленное множество решений. Из них интерес представляют лишь такие значения, для которых найденные значения ψ2 (плотность вероятности) не противоречат физическим представлениям. Таковыми являются квантовые числа n, l, ml, s.
§
-Не связано с движением электрона вокруг ядра;
-Характеризует собственный момент вращения электрона вокруг своей оси;
-Значения: 1/2(движение электрона по часовой стрелке); —1/2(против).
Голландские физики Уленбек и Гоудсмит открыли спин электрона в 1925 г. Спин это не просто вращение электрона как «волчка», а сложное физическое явление. Дирак в 1928 г. показал наличие параллельных () и антипараллельных (¯) спинов.
Для определения состояния электрона в атоме важное значение имют: принцип минимума энергии, определяющий заполнение АО с наименьшей энергией (1s<2s<2p<3s<4s»3d<4p<5s»4d<5p<6s»4f»5d<6p<7s); принцип Паули, диктующий присутствие на АО не более 2-х электронов, с противоположно направленными спинами, и правило Хунда, предписывающее заполнение АО электронами так, чтобы их суммарный спин был максимальным.
ПРИНЦИП ПАУЛИ — В атоме не может быть двух электронов, у которых все четыре квантовых числа были бы одинаковы (1925 г). Попробуем «создать» электронные оболочки атомов, пользуясь принципом Паули. Минимальное значение главного квантового числа n равно 1. Ему соответствует только одно значение орбитального квантовогочисла l равное нулю (s-орбиталь).Сферическая симметрия s-орбиталей выражается в том, что при l=0 в магнитном поле существует только одна орбиталь с ml=0 . На этой орбитали может находиться один электрон с любым значением спина (водород, его электронная формула 1s1) или два  с противоположным значением спинов (гелий, его электронная формула 1s2). Таким образом, при значении n=1 может существовать на первом энергетическом уровне не более двух электронов.
с противоположным значением спинов (гелий, его электронная формула 1s2). Таким образом, при значении n=1 может существовать на первом энергетическом уровне не более двух электронов.
Теперь начнем заполнять орбитали с n=2 (на первом уровне уже есть два электрона). Величина n=2 соответствуют два значения орбитального числа: 0(s-орбиталь ) и 1(р-орбиталь ). При l=0 существует одна орбиталь,при l=1-три орбитали(со значениями ml:-1, 0, 1) .На каждой из орбиталей может находиться не более двух электронов, так что значению n=2 соответствует максимум 8 электронов . Общее число электронов на уровне с заданным n можно вычислить , таким образом , по формуле 2n².(Далее смотрите табл.№3,№4).
ПРАВИЛО ХУНДА — устойчивому состоянию атома соответствует такое распределение электронов в пределах энергетического подуровня, при котором абсолютное значение суммарного спина атома |S| максимально.Устойчивое состояние атома — это состояние с максимальным числом неспаренных электронов. Например: электронное строение атома углерода 6С 2s²2p² может быть описно различными вариантами электронно-графических формул а-в.
Запрещено :
| а)2р | ↑↓ | |S|=0 | б)2р | ↑ | ↓ | | S|=0 |
Разрешено:
Состояние в) более устойчиво так как абсолютное значение его суммарного спина |S| максимально.
§
Клечковским.
1-е ПРАВИЛО КЛЕЧКОВСКОГО: При увеличении заряда ядра атома последовательное заполнение электронных орбиталей происходит от орбиталей с меньшим значением суммы (n l) к орбиталям с большим значением этой суммы.
Например, атом калия, электронная формула которого 1s2 2s2 p6 3s2 p6 d 0 4s1 на третьем энергетическом уровне имеет только восемь электронов, из 18-ти возможных. Почему электрон заполняет 4s, а не 3d – подуровень? Рассчитавдля атома калия по правилу Клечковского сумму (n l) для Зd(3 2=5) и 4s(4 0=4) подуровней, делаем вывод, что в первую очередь должен заполнятся 4s, а потом 3d-подуровень, в соответствии с первым правилом Клечковского.
2-е ПРАВИЛО КЛЕЧКОВСКОГО: Если у орбиталей сумма главного квантового числа и орбитального квантового числа (n l) одинакова, то заполняется прежде орбиталь с меньшим значением главного квантового числа n.
При заполнении электронных орбиталей у атома скандия, электронная формула которого 1s22s2p63s2p6d14s2 , электрон заполняет 3d(3 2=5), а не 4р(4 1=5)-подуровень, в соответствии со вторым правилом Клечковского.
У некоторых атомов наблюдается явление «электронного проскока», как, например, у атома хрома: Cr 3d44s2→3d54s1 или у атома меди: Cu3d94s2→3d104s1.
Это объясняется повышенной энергетической устойчивостью электронных конфигураций с полностью (атом Сu) или наполовину заполненным подуровнем (атом Cr ).
ПЕРИОДИЧЕСКИЙ ЗАКОН Д.И.МЕНДЕЛЕЕВА.
ЗАКОНОМЕРНОСТИ ПЕРИОДИЧЕСКОЙ СИСТЕМЫ.
План
1. Периодический закон Д.И.Менделеева.
2. Строение периодической системы Д.И.Менделеева.
3. Физический смысл величин периодической системы.
4. Закономерности периодической системы. Радиус атома. Валентность элемента.
5. Энергия ионизации, энергия сродства к электрону, электроотрицательность.
6. План характеристики свойств элементов по их положению в периодической системе.
Литература:
1. Фролов В.В. Химия. Гл.V, §51-56.
2. Лучинский Г.П. Курс химии. Гл.V, §8-12, гл. VI, §13-18
3. Ахметов Н.С. Общая и неорганическая химия. Раздел V, гл.3,4.
4. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
Периодический закон был сформулирован 1 марта 1869 года великим русским ученым Дмитрием Ивановичем Менделеевым.
Периодический закон в интерпретации Д.И.Менделеева: свойства простых тел, а также формы и свойства их соединений находятся в периодической зависимости от величины атомных весов элементов.
Современная формулировка периодического закона: свойства простых тел, а также формы и свойства их соединений находятся в периодической зависимости от величины зарядов их атомных ядер.
Периодическая система элементов является графическим изображением периодического закона. Известно несколько сотен форм периодической системы, однако, на Украине чаще используется короткопериодный вариант периодической системы. Периодическая система состоит из периодов и групп.
Период – это горизонтальный ряд элементов, расположенных в порядке возрастания порядкового номера от первого s-элемента (ns1) до р-элемента (ns2np6). Каждый период (кроме первого) начинается активным щелочным металлом и заканчивается инертным газом, перед которым стоит активный не металл (галоген). В периодах происходит заполнение электронами внешней (n) электронной оболочки атомов, а также незаполненных предвнешних (n-1) и (n-2) оболочек. Различают: малые (1,2,3) и большие (4,5,6,7) периоды. В периоде слева направо металлические свойства элементов убывают, а неметаллические – возрастают.
Номер периода указывает на число энергетических уровней (электронных оболочек) в атоме и совпадает со значением главного квантового числа (n) внешнего энергетического уровня.
Группы – это вертикальный ряд элементов, обладающих однотипным электронным строением. В соответствии с максимальным числом электронов на внешнем электронном слое невозбужденных атомов элементы периодической системы подразделяются на восемь групп. В группе выделяют главную (А) (в нее входят s- и р — элементы малых и больших периодов) и побочную (В) (в нее входят элементы только больших периодов) подгруппы.
Номер группы соответствует высшей валентности элемента.
Под валентностью химического элемента подразумевается его способность к образованию химических связей. В представлении метода валентных связей численное значение валентности соответствует количеству неспаренных электронов. В периодической системе каждый элемент имеет строго определенный порядковый номер и занимает строго определенное место.
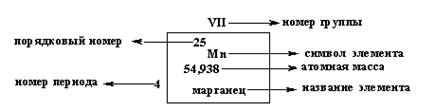
Порядковый номер элемента соответствует положительному заряду ядра атома (количеству протонов — ∑ ) и общему количеству электронов (  ) на его орбиталях.
) на его орбиталях.
Валентные электроны обладают наибольшим запасом энергии, они определяют химические свойства атомов. Атомы, вступая в химические реакции, могут отдавать валентные электроны, принимать их или образовывать общие электронные пары.
Способность атомов отдавать или принимать электроны определяется радиусом атома (Rат) хотя эта величина физическая, в ней заключен глубокий химический смысл. Чем больше Rат, тем легче атом теряет свои валентные электроны и труднее их присоединяет.
В периоде Rат уменьшается слева направо, так как с увеличением порядкового номера элемента количество энергетических слоев или оболочек не меняется, а количество электронов на них и заряд ядра атома увеличиваются. Это приводит к сжатию электронной плотности и как следствие, уменьшению Rат.
В группе Rат увеличивается сверху вниз, так как с увеличением порядкового номера элемента, увеличивается количество оболочек и силы, связывающие электроны с положительно заряженным ядром атома ослабевают. Атомы, отдающие электроны, проявляют восстановительныесвойства.
Количественной характеристикой восстановительных свойств элементов являетсяэнергия ионизации (I , кДж/моль, эВ/атом).
Энергия ионизации – это количество энергии, необходимое для отрыва электрона от невозбужденного атома с превращением последнего в положительно заряженный ион (катион). Первая энергия ионизации характеризует способность атома отдавать один электрон: Э0 I 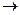 Э ē.
Э ē.
Для многоэлектронных атомов энергии ионизации I1; I2; I3… соотвествуют отрыву первого, второго и т.д. электронов. При этом I1< I2 <I3…, т.к. увеличение числа оторванных электронов приводит к возрастанию положительного заряда образующегося иона.
Отношение энергии ионизации к заряду электрона называется потенциалом ионизации (измеряется в электрон-вольтах, (эВ)). Потенциал ионизации численно равен энергии ионизации.
В периоде значение энергии ионизации от щелочных металлов к инертным газам увеличивается, т.к. Rатуменьшается, а заряд ядра атома увеличивается в результате чего происходит сжатие электронной плотности, поэтому необходимо затрачивать большее количество энергии для отрыва электрона от нейтрального атома.
В группах за счет увеличения радиуса атома сверху вниз, энергия ионизации уменьшается.Самая малая энергия ионизации у франция 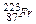 .
.
Атомы, принимающие электроны, проявляют окислительныесвойства. Количественной характеристикой окислительных свойств элемента является энергия сродства к электрону (Е; эВ/атом).
Энергия сродства к электрону – это энергия, выделяющаяся при присоединении электрона к нейтральному атому, она характеризует способность атома к образованию отрицательно заряженных ионов: Э0 ē  Э— Е.
Э— Е.
Наибольшее сродство к электрону у атома F. Энергия сродства к электрону значительно меньше энергии ионизации тех же атомов.
Обе эти величины (I и Е) зависят от заряда ядра и размеров атома: с увеличением заряда ядра они должны расти, а с увеличением радиуса атома – уменьшаться.
Для сравнительной оценки I и Е в 1932 году американским ученым Полингом введена особая характеристика – электроотрицательность (ЭО), которая определяется по Малликену, как способность атомов в молекуле притягивать электронную плотность.
ЭО = 1/2(I Е).
Чем больше величина ЭО, тем сильнее выражены у элемента неметаллические свойства. Чем меньше величина ЭО, тем сильнее выражены — металлические свойства. Эта величина не имеет строго физического смысла, но удобна для оценки реального состояния химической связи в молекулах. За единицу ЭО принята ЭО лития. Самым электроотрицательным элементом является фтор.
С точки зрения строения атома все элементы можно разделить на четыре типа:
s — элементы (заполняется s- подуровень АО внешней оболочки): …ns1, ns2
p — элементы (заполняется p -подуровень АО внешней оболочки) … np1— np6.
d — элементы (заполняется d-подуровень АО незаполненной предвнешней оболочки) … (n-1)d1ns2 – (n-1)d10ns1.
f — элементы (заполняется f-подуровень АО незаполненной третьей снаружи оболочки) … (n-2)f1 ns2 — (n-2)f14 ns2.
Так как на s – подуровне может быть (1-2) е, то s-элементы располагаются в IА и IIА группах, главных подгруппах. Это типичные металлы, имеют сходные химические свойства,так как являются электронными аналогами.
Электронные аналоги – это элементы, у которых валентные электроны расположены на орбиталях, описываемых общей для всех электронной формулой:
Li Na K Pb Cs Fr
2s¹ 3s¹ 4s¹ 5s¹ 6s¹ 7s¹
ns1 – электронные аналоги
Таблица №5
Be Mg Ca Sr Ba
2s2 3s2 4s2 5s2 6s2
ns² – электронные аналоги
Таблица №6
На p – подуровне может быть (1-6) электронов, поэтому p – элементы располагаются в IIIA – VIIIA группах, главных подгруппах. Таким образом, главные подгруппы составляют только s- и p – элементы. Среди p–элементов могут быть металлы, но они чаще всего амфотерны:
Таблица №7
p–элементы у которых (4-8) электронов на последнем энергетическом уровне являются неметаллами.
Таблица №8
17Cl (проявляет переменную валентность за счет появления вакантного 3d – подуровня, на который могут переходить валентные электроны с 3s и 3p подуровней).
Таблица №9
У инертных газов (He и Ne) p–подуровень полностью заполнен электронами. Теоретически и практически распариваться электронам некуда. Валентность равна нулю (He;Ne).
У 18Ar за счет появления вакантного 3d – подуровня возможны валентности:
Внорм=0; В*=2,4,6,8.
54Xe горит во фторе с образованием: XeF2, XeF4, XeF6, XeF8.
d – элементы — металлы ,так как имеют на последнем электронном уровне 2 электрона, хотя заполнение вакантных орбиталей у них происходит на предвнешнем слое. Их называют переходными, по расположению в периодической системе между s и p элементами, они формируют побочные группы.
Таблица №10
План характеристики свойств элементов по положению в периодической системе (ПС).
1. Положение элемента в ПС (порядковый номер, период, группа, подгруппа).
2. Строение атома (заряд ядра и его состав) – число протонов, нейтронов, число электронов в атоме, структура электронной оболочки атома, электронная формула валентных уровней, валентность в основном и возбужденных состояниях.
3. Тип элемента (s-, p-, d-, f- элемент), (металл, неметалл), возможные степени окисления, формулы оксидов и гидроксидов для каждого валентного состояния.
Пример: дать характеристику натрия. Порядковый номер его в периодической системе 11, следовательно, заряд ядра атома равен 11, вокруг ядра вращаются 11 электронов. Натрий расположен в третьем периоде, поэтому его электроны располагаются на трех энергетических уровнях. Натрий расположен в I — ой группе, главной подгруппе, следовательно, у атома Na один валентный электрон на внешнем уровне. А если у атома на внешнем энергетическом уровне находится 1,2,3 электрона, то атомы таких элементов являются металлами. Следовательно, Na – металл, его оксид Na2O имеет основной характер. Ему соответствует основание NaOH.
Открытие периодического закона Д.И. Менделеевым и создание им периодической системы химических элементов явились триумфом в развитии химии XIX столетия.
Накопившиеся к тому времени знания о свойствах 63 химических элементах были приведены Д.И. Менделеевым в строгий порядок. С открытием периодического закона появилась возможность предвидеть свойства элементов и их соединений. Д.И.Менделеев на основе открытого им периодического закона предсказал свойства тогда ещё неизвестных элементов: Sc, Ga, Ge, Tc, Re, Po, At, Fr, Ra, Ac, Pa., смог исправить атомные веса уже известных в то время элементов: Be, Ti, Y, In, La.
Периодическая система элементов оказала большое влияние на последующее развитие химии. Она не только была первой естественной классификацией химических элементов, показавшей, что они образуют стройную систему и находятся в тесной связи друг с другом, но и явилась могучим орудием для дальнейших исследований. С течением времени предсказания Д.И.Менделеева блестяще подтвердились. Все предсказанные элементы были открыты и их свойства с поразительной точностью соответствовали ранее описанным.
В настоящее время периодический закон остается руководящим принципом химии. Именно на его основе искусственно создают трансурановые элементы. Один из них – элемент №101, впервые полученный в 1955г. – в честь великого русского ученого был назаван Менделевием.
ХИМИЧЕСКАЯ ТЕРМОДИНАМИКА.
План
1. Определение понятия « химическая термодинамика».
2. Основные понятия раздела «химическая термодинамика»:
а) термодинамическая система, виды систем;
б) термодинамические параметры;
в) процесс, классификация процессов;
г) цикл;
д) термодинамические функции.
3. Первое начало термодинамики:
а) изохорный процесс, внутренняя энергия системы
б) изобарный процесс, определение понятия «энтальпия».
4. Термохимия:
а) определение понятия «термохимия»;
б) тепловой эффект химической реакции;
в) основные законы термохимии, закон Гесса.
Литература:
1. Фролов В.В. Химия. Гл.V, §51-56.
2. Лучинский Г.П. Курс химии. Гл.V, §8-12, гл. VI, §13-18
3. Ахметов Н.С. Общая и неорганическая химия. Раздел V, гл.3,4.
4. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
Термодинамика – как наука возникла в начале XIX в. (в связи с задачами совершенствования тепловых машин). В переводе с греческого «термос» означает тепло, а «динамос» – силу и мощь. Классическая термодинамика занимается исследованием энергии и работы в макроскопических системах. Этим объясняется ее значение для таких наук, как физика, химия, биология, геология, для многочисленных отраслей техники, так как любые процессы, происходящие в природе, сопровождаются изменениями энергии.
Химическая термодинамика – это раздел химии, который
изучает переходы энергии из одной формы в другую при химических процессах и устанавливает направление и пределы их самопроизвольного протекания при данных условиях.
Термодинамическая химическая система – комплекс взаимодействующих между собой веществ, мысленно обособленный от окружающей среды. Например, системой может быть химический стакан, содержащий определенное количество воды, либо теплообменник, используемый на химическом предприятии и т.д.
Различают три типа термодинамических систем (табл.11).
Изолированныесистемы – не могут обмениваться с окружающей средой ни энергией, ни массой. Например, изолированный термостат, Вселенная в целом.
Закрытыесистемы – могут обмениваться с окружающей средой только энергией, но не массой. Например, совокупность молекул растворенного вещества можно рассматривать как закрытую систему, а в качестве внешней среды может быть все остальное (возможно растворитель, если он не участвует в реакции). Поэтому в химической термодинамике наиболее часто рассматривают именно закрытые системы.
Открытыесистемы – это системы, которые могут обмениваться с окружающей средой и энергией, и массой. Например, живые объекты животного или растительного мира.
Таблица №11
Какую бы систему мы не рассматривали, она может быть в различных состояниях. А чтобы описать то или иное состояние, используют термодинамические характеристики (рис.4). Одни термодинамические характеристики можно рассматривать как основные, которые определяют состояние вещества. Их называют параметрами состояния. Обычно это температура (Т), давление (Р) и количество вещества (n).
Остальные термодинамические характеристики зависят от этих трех параметров (Т, Р, n), а значит в целом и от состояния системы. Поэтому их называют функциями состояния.
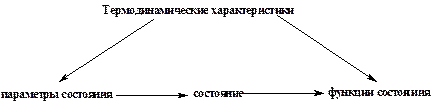
Рис.4
Термодинамический процесс – это всякое изменение параметров состояния (т.е. переход системы из одного состояния в другое). Процессы бывают:
— изохорный (V=const);
— изобарный (P=const);
— изотермический (T=const);
— адиабатный (нет обмена энергией системы с окружающей средой);
— изобарно — изотермический (P;Т=const);
— изохорно — изотермический (V;T=const).
Первое начало термодинамики было сформулировано Джоулем в середине ХIX в. По своей сути это закон сохранения энергии в применении к термодинамическим системам: тепло переданное системе (Q) идет на изменение ее внутренней энергии (DU) и на совершение работы (А): Q=DU A
Внутренняя энергия системы (U) – это сумма энергии теплового движения молекул, внутримолекулярной энергии и энергии межмолекулярного взаимодействия.
Абсолютное значение этой величины не известно. Поэтому оперируют лишь величинами DU – изменениями внутренней энергии в процессах. DU не зависит от способа перехода системы из одного состояния в другое. Внутренняя энергия является функцией состояния системы.
Что касается теплоты (Q) и работы (А), то это две единственные формы передачи энергии от системы к внешней среде и обратно. Знаки теплоты и работы определяются схемой (рис.5).
1.Q>0; А>0, если система получает теплоту или над ней совершается работа.
2.Q <0; А<0, если система отдает теплоту или сама производит работу.
Соответствующий знак (плюс или минус) ставится не перед буквами Q и А, а перед цифровым значением соответствующей величины, например, Q=-300 кДж или А= 50 кДж.



 Q>0
Q>0
A>0
Рис.5
В общем случае ни работа, ни теплота не являются функциями состояния.
Первое начало термодинамики для различных процессов в системе идеального газа.
Изотермические процессы идут при постоянной температуре (T=const).
Для их осуществления расширение или сжатие газа надо сочетать с подведением или забором теплоты так, чтобы температура газа оставалась постоянной. Согласно выражению энергии для идеального газа: U=3/2nRT, внутренняя энергия идеального газа зависит только от температуры. Поэтому при постоянной температуре DU=0, и выражение первого закона термодинамики примет вид: QT = AT.
Таким образом, при изотермическом расширении газа вся подводимая извне теплота трансформируется в работу газа, а при сжатии газа отводимая теплота равна работе, совершаемой над газом.
Адиабатиые процессы – это процессы, при которых исключен теплообмен системы с окружающей средой (Q=0), откуда A= DU. В этих случаях работа становится функцией состояния.
Изохорные процессы (V=const).
Например, нагревание газа под фиксированным поршнем. Естественно, что работа при этом не совершается (А=0), откуда QV = DU.
1.Теплота при изохорных процессах (QV) является функцией состояния системы, т.к. таковой является равная ей величина DU.
2.Это первое математическое выражение закона Гесса (см. далее).
Изобарные процессы (P=const).
Например, нагретый газ совершает работу расширения против постоянного внешнего давления. Работа расширения (АР) определяется как произведение давления (Р) на изменение объема (DV). АР=РDV, откуда: QP=DU PDV
1.Теплота изобарного процесса (QP) является функцией состояния системы, так как работа против постоянного внешнего давления не зависит от способа совершения процесса, она определяется только начальным и конечным состоянием системы DV. Следовательно, работа является функцией состояния, если в ходе изобарного процесса не совершается никакой иной работы, как против постоянного внешнего давления.
Обозначим сумму (U2 PV2) через величину Н2, а (U1 PV1) = H1, тогда QP= H2— H1= DH. Таким образом, в термодинамику введена новая величина Н — энтальпия. Особенность энтальпии состоит в том, что она является функцией состояния. Следовательно, DH= H2 — H1, во всех процессах (как и DU) зависит только от начального (Н1) и конечного (Н2) состояний.
2.Выражение QP = DH это второе математическое выражение закона
Гесса (см. далее).
QP = U2 – U1 P (V2 – V1) = (U2 PV2) – (U1 PV1) = Н2 – Н1 =DH.
Для экзотермических реакций QV > 0, тепло система отдает в окружающую среду, значит, ее внутренняя энергия уменьшается Þ DН < 0.
§
Закон Гесса является основным законом термохимии (1840 г.):
Тепловой эффект химической реакции не зависит от механизма протекания реакции, а зависит лишь от природы и физического состояния исходных веществ и продуктов реакции (при условии V=const; P=const).
Проиллюстрируем данное утверждение на примере.
Димер оксида азота (IV) может быть получен несколькими способами.
Во-первых, 1) N2 O2 = 2NO, DH1= 180900 Дж/моль;
2) 2NO O2 = 2NO2, DH2= -77100 Дж/моль;
3) 2NO2 = N2O4, DH3=-10800 Дж/моль.
Или, во-вторых,
4) N2 2O2 = N2O4, DH4=93000 Дж/моль.
DH1 DH2 DH3 = DH4.
Более наглядно графическое выражение этих процессов:
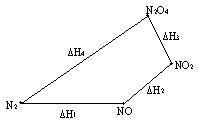
Рис.6
Поскольку в определение энтальпии входит внутренняя энергия, то абсолютное значение энтальпии системы (или какого-либо вещества) неизвестно. Поэтому энтальпию вещества характеризуют энтальпиями образования и сгорания.
DН0298(обр) (стандартная энтальпия образования) – это теплота реакции образования вещества (Х) из простых веществ (или элементов) при стандартных условиях: простые вещества ® Х.
Для простых веществ DН0обр равно нулю (в устойчивых агрегатных состояниях).
DН0сг (стандартная энтальпия сгорания) – теплота реакции сгорания вещества до оксидов (с максимальной степенью окисления) при стандартных условиях: Х О2 ® оксиды (с максимальной степенью окисления).
Значения DН0обр и DН0сг содержатся в справочных таблицах.
Энтальпии многих реакций не поддаются экспериментальному определению, так как их проведение в лабораторных условиях невозможно. Например, в лабораторных условиях невозможно определить энтальпию образования этанола, так как его невозможно синтезировать из атомов С, Н, О. Энтальпии подобных реакций могут быть вычислены по известным энтальпиям других реакций с помощью одного из следствий закона Гесса.
Следствия из закона Гесса:
Тепловой эффект химической реакции может быть найден, как разность между суммой теплот образования продуктов реакции и суммой теплот образования исходных веществ, с учетом стехиометрических коэффициентов.
DНх.р. = SDН0 .298(обр) продуктов реакции – SDН0 298(обр) исходных веществ, с учетом стехиометрических коэффициентов.
Тепловой эффект химической реакции может быть найден, как разность между суммой теплот сгорания исходных веществ и суммой теплот сгорания продуктов реакции, с учетом стехиометрических коэффициентов.
DНх..р. = SDН0298(сгор) исходных веществ– SН0298(сгор) продуктов реакции с учетом стехиометрических коэффициентов.
S — греческая буква сигма, обозначающая математическую операцию суммирования.
Задача: Определить стандартную энтальпию образования DН0обр (РН3), исходя из уравнения.
2РН3(г) 4О2(г) = Р2О5(к) 3Н2О (ж), DНх.р = -2360 кДж.
Решение:
DНх. р. = [DН0обр (Р2О5(к)) 3DН0обр (Н2О (ж))] — 2DН0обр (РН3);
2DН0обр (РН3)= [DН0обр (Р2О5(к)) 3DН0обр (Н2О (ж))] — DН0х. р.,
DН0обр (Р2О5(к)) = -1492 кДж/моль, DН0обр (Н2О (ж)) = -285,8 кДж/моль.
2DН0обр (РН3)= [(-1492 3(-285,8))] – (-2360)= 10.6 кДж/моль.
DН0обр (РН3)= 10,6/2=5,3 кДж/моль.
(т) — твердое агрегатное состояние, (ж)-жидкое агрегатное состояние, (г)- газообразное агрегатное состояние.
Не только химические, но и физико-химические процессы могут сопровождаться выделением или поглощением теплоты.
Растворение веществ.
Если вещество находится в твердом состоянии, то процесс можно представить в две стадии:
Вещество с кристаллической структурой 
DН0кр
молекулы  сольватированные молекулы
сольватированные молекулы
DН0сольв.
На первой стадии разрушается кристаллическая решетка, обычно это эндотермический процесс (DН0кр>0). На второй – происходит сольватация (гидратация, если растворитель вода) молекул вещества, это экзотермический процесс (DН0сольв<0). Результирующая теплота растворения DН0раств = DН0кр DН0сольв может иметь как положительный, так и отрицательный знак.
При растворении газов отсутствует стадия разрушения кристаллической решетки. Остается экзотермическая сольватация. Поэтому растворение газов, как правило, экзотермично.
При растворении кристаллогидратов отсутствует стадия сольватации. Остается лишь эндотермическое разрушение кристаллической решетки. Процесс идет с поглощением теплоты.
В качестве примера рассмотрим:
1.Растворение безводной соли CuSO4 (т) сульфата меди:
CuSO4(т) ® CuSO4(ж)® CuSO4. 5 H2O.
DН0кр = 11,7 кДж/моль; DН0сольв = -78,2 кДж/моль;
DН0раств = 11,7 – 78,2 = -66,5 кДж/моль
2.Растворение кристаллогидрата CuSO4. 5 H2O
1. CuSO4. 5 H2O (т) ® CuSO4. 5 H2O (ж)
DН0раств = DН0кр = 11,7 кДж/моль.
Реакция нейтрализации.
Сильные кислоты и основания в растворе диссоциированы полностью, и поэтому теплота реакции нейтрализации (не зависимо от природы реагирующих веществ) одна и та же.
Рассчитаем ее:
Н (ж) ОН— (ж) ® Н2О (ж)
DН0обр. кДж/моль 0 -230 -285.8
DН0нейтр. = -285.8 230 = -55.8 кДж/моль.
Если же в подобной реакции участвует слабый электролит, то процесс можно записать в две стадии:
ОН—
НА ® Н А— ® А — Н2О
DН0дис. DН0нейтр.
Первая стадия – это ионизация слабого электролита. Она обычно является эндотермической, когда для полной ионизации всех молекул слабого электролита требуется передать веществу теплоту. И чем слабее кислота, тем больше надо теплоты.
Итак, в реакции нейтрализации, идущей с участием слабого электролита, результирующее выделение энергии оказывается меньше, чем в случае сильного электролита.
§
Теплота фазового перехода равна разности DН0обр вещества в одном и в другом состояниях. Для испарения воды (250С) имеем:
Н2О(ж)®Н2О(г) DН0обр. кДж/моль -285.8 -241.8
DНисп.= -241.8 285.8= 44 кДж/моль (эндотермический процесс).
ВТОРОЕ НАЧАЛО ХИМИЧЕСКОЙ ТЕРМОДИНАМИКИ.
План
1. Понятие термодинамической вероятности.
2. Энтропия.
3. Формулировки II закона термодинамики.
4. Энергия Гельмгольца.
5. Энергия Гиббса.
Литература:
1. Фролов В.В. Химия. Гл.VI, §6.1 – 6.11.
2. Лучинский Г.П. Курс химии. Гл. IV, §1-7.
3. Ахметов Н.С. Общая и неорганическая химия. Раздел V, гл. 1,2.
4. Общая химия под ред. Соколовской Е.М. и др. Гл. 5, § 1-6.
Первый закон термодинамики, с которым вы познакомились при рассмотрении предыдущей лекции, по своей сущности является законом сохранения. Он позволяет рассчитывать энергетику процесса, однако не решает вопрос возможности протекания процесса и если процесс возможен, то о его направлении. Например, протекание таких процессов как самопроизвольное разделение газов или переход тепла от холодного тела к горячему невозможно, так как в природе все процессы протекают сами собой с определенной направленностью, т.е. самопроизвольно теплота может переходить от горячего тела к холодному и т.д., что является следствием молекулярной природы вещества. Однако, такие не реальные явления, как самопроизвольное разделение газов или переход тепла от холодного тела к горячему не противоречат первому закону термодинамики.
Рассмотрим одностороннее протекание процессов на примере выравнивания температур между частями системы. Это переход менее вероятного упорядоченного состояния (рис.7а): частицы с большей энергией (Т2; Т2 > Т1) находятся в одной части пространства, а частицы с меньшей энергией – в другой, в более вероятное состояние беспорядка (рис.7 б) (Т1=Т2=Тср).
Рис. 7а Рис. 7 б
Или, например, допустим, что все молекулы вещества сосредоточены в небольшой части V1 объема V0 (рис.8а). Это означает наличие в системе высокой энергетической упорядоченности, так как молекулы являются носителями внутренней энергии. Из опыта известно, что такой порядок неустойчив, менее вероятен: частицы (если они ничем не зафиксированы) стремятся занять весь объем V0 (рис.8б). В результате в системе повышается степень энергетического беспорядка, т.е. состояние системы (рис.8б) более вероятно.
Рис. 8а Рис.8б
Для характеристики более вероятных процессов (рис.7б; 8б) в термодинамике используют понятие – термодинамическая вероятность, W.Термодинамическая вероятность, W – это количество микросостояний системы.
§
«Коэффициент полезного действия паровой машины всегда меньше единицы» и т.д.
Но они, в большей степени, важны для физических процессов. А для химических процессов важно то, что второе начало термодинамики дает критерии того, какие процессы могут проходить самопроизвольно, а какие нет.
Опыт показывает, что многие самопроизвольно протекающиепроцессы идут с выделением теплоты (DН<0). Но среди самопроизвольных процессов встречаются также эндотермические (DН>0). Примером является растворение аммония нитрата в воде:
NH4NO3 (т) ® NH4NO3 (ж); DН0х.р. = 27 кДж/моль.
Получается, тепловыделение не является решающим критериемвозможности самопроизвольного протекания процессов.
Так же могут самопроизвольно протекать процессы с уменьшением энтропии (DS<0): С О2 ® СО2.
Между молекулами углерода С и кислорода О2 устанавливается связь, и поэтому степень энергетического беспорядка уменьшается.
Каков же критерий самопроизвольности – теплота или энтропия?
Критерием, определяющим возможность протекания процесса, есть баланс действия энтальпии (Н) и энтропии (S).Поэтому возможны самопроизвольные процессы с поглощением теплоты (если рост энергетического «беспорядка» является более весомым), и процессы с DS<0, (если это сопровождается более сильным выделением теплоты).
Измеряется изменение энтропии приведенной теплотой обратимого процесса, связывающего соответствующие состояния системы:
DS = Qобр./Т.
Для необратимых процессов: DS >Qнеобр./Т (алгебраическая сумма приведенных теплот для необратимых процессов: Qнеобр.проц. < Qобр .проц.
Математическая формулировка II закона термодинамики (обобщенная формула) выглядит следующими образом:
DS ³ Q/T
Для изолированных систем, где обмен энергией с окружающей средой исключен Q=0, неравенство, выражающее второе начало термодинамики примет вид: DS ³0.
Таким образом, для изолированных систем остается только один критерий самопроизвольного протекания процессов – увеличение энтропии. Самопроизвольными являются лишь процессы, ведущие к увеличению общей энтропии.Итак:
1.Энтропия – это функция состояния системы, поэтому вычислять изменение энтропии можно, используя следствие из закона Гесса. Энтропия химической реакции равна разности между суммами энтропий продуктов реакции и энтропий исходных веществ, с учетом стехиометрических коэффициентов.
DSх.р. = S Sпрод. — S Sисх.в-в, с учетом стехиометрических коэффициентов.
Задача: Рассчитать изменение энтропии химических реакций (DSх.р.) для процесса: 3С2Н2 (г) ® С6Н6 (ж).
Решение: DSх.р. = S0 (C6H6 (ж)) – 3 S0 (С2Н2 (г)).
DSх.р. = 269,2 – 3 . 200,8 = — 333,2 Дж/моль.
2.Энтропия измеряется приведенной теплотой.
Задача: Чему равно приращение энтропии моля Fe при Тпл=15360С (1809 К) при переходе из кристаллического в жидкое состояние, если энтальпия DНпл = 13765 Дж/моль.
При температуре плавления Feкр Û Feж ( в равн. Þ T=const) = Тпл.
ß
DSпл = S (Fe (ж)) -S (Fe (т)) =DНпл/Тпл= 13765/1809= 7,61 Дж/К. моль.
DS>0 Þ процесс при Тпл протекает самопроизвольно.
3.Энтропия является критерием самопроизвольного протекания процессов только для изолированных систем. Самопроизвольно протекают в изолированных системах только те процессы, которые идут с увеличением энтропии.
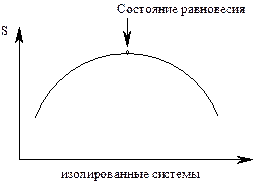
DS³0 DS³0
рис 9
4. S связана с числом возможных микросостояний в системе (, т.е. с термодинамической вероятностью,W), характеризует меру энергетического беспорядка. S= klnW в системе.
5. S – это термодинамическая функция, которая в отличие от термодинамической вероятности W связана с термодинамическими величинами Q; T, т.е. более приемлема для химической термодинамики.
Однако, в природных процессах и в технике чаще встречаются системы взаимодействующие с окружающей средой, т.е. неизолированные. Энтропия, S мало подходит для характеристики процессов в этих условиях.
Для изохороно-изотермических условий первый закон термодинамики имеет следующую математическую форму записи:
QV = DU (A= РDV; DV= const Þ A=0)
Из II –го закона термодинамики следует: DS ³ QV /T Þ QV £ ТDS.
Объединяя два выражения, получим: TDS ³ DU;DU — TDS £ 0.
Обозначим разность DU — TDS через новую термодинамическую функцию состояния – свободную энергию Гельмгольца, DU — TDS =DF.
Следовательно II-е начало термодинамики применительно к изохороно-изотермическим процессам можно записать так: DF£ 0.
Введение данной функции позволяет пользоваться лишь одним критерием направления процесса (какой для изолированных систем является S). Однако этот чисто формальный прием не отменяет того факта, что на самом деле для самопроизвольных процессов по прежнему остаются те же критерии: DН и DS. Через DF обозначен их баланс.
Итак: 1. для изохорно-изотермических условий DF является
суммарным критерием самопроизвольного протекания процессов
DF=DU — TDS (это выражение объединенных I и II законов термодинамики).
2.Самопроизвольными являются такие процессы для изохорно изотермических условий, в которых энергия Гельмгольца убывает
DF< 0).
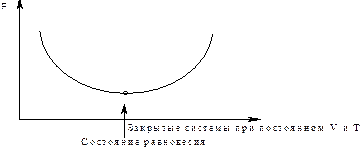
DF£ 0 DF£ 0
DF= 0
рис10
DF<0, процессы могут идти самопроизвольно; DF= 0, процессы занимают пограничное положение, характеризующееся минимальным значением;
DF > 0, процессы сами проходить не могут.
3.Энергия Гельмгольца является функцией состояния системы, поэтому вычислить изменение энергии Гельмгольца можно, используя следствие из закона Гесса.
Энергия Гельмгольца химической реакции равна разности между суммами энергий Гельмгольца продуктов реакции и энергий Гельмгольца исходных веществ, с учетом стехиометрических коэффициентов.
DFх.р. = SDF0прод. — SDF0исх.в-в, с учетом стехиометрических коэффициентов.
Для изобарно-изотермических условийI-ый закон термодинамики можно записать в виде: QP = DU РDV; т.е.теплота при изобарно-изотермических условиях равна изменению энтальпии QP = DU pDV = DH (при условии, что не совершается никакой иной работы, как против давления). Объединяя неравенство D S ³ QP/T (выражение II закона термодинамики), получим вид: DН — ТDS £0. Если выражение DН — ТDS обозначить через новую термодинамическую функцию состояния – свободную энергию Гиббса, DG :
DН — ТDS = DG,
следовательно II –е начало термодинамики, применительно к изобарно-изотермическим процессам можно записать так: DG £0.G – энергия Гиббса, является также как и энергия Гельмгольца лишь балансом все тех же двух критериев самопроизвольного протекания процессов: выделения теплоты и возрастания энтропии. Итак,
1. для изобарно-изотермических условий DG является суммарным критерием самопроизвольного протекания процессов DG = DН — ТDS ( объединенный ( I и II ) закон термодинамики).
2.Процессы, в которых DG убывает (DG< 0) протекают самопроизвольно.
D G < 0, химические процессы могут идти самопроизвольно;
DG = 0, химические процессы занимают пограничное (равновесное) положение;
DG > 0, химические процессы сами проходить не могут.
DG £ 0 DG £ 0
DG = 0
рис.11
3. Энергия Гиббса DG является функцией состояния системы, поэтому вычислить изменение энергии Гиббса химических реакций можно по следствию из закона Гесса. Энергия Гиббса химических реакций равна разности между суммами энергий Гиббса продуктов реакции и энергий Гиббса исходных веществ, с учетом стехиометрических коэффициентов.
DGх.р= SDG0прод. — S DG0исх..в-в., с учетом стехиометрических коэффициентов.
DG0обр. (Х) – стандартная энергия Гиббса вещества Х при стандартных условиях и в определенном агрегатном состоянии.
DG0сг. (Х) – стандартная энергия сгорания вещества – это DG реакции окисления до конечных оксидов 1 моля Х. Для простых веществ и элементов DG0обр.(Х) (как и DН0обр.(Х)) равны нулю.
Задача: Спиртовое брожение происходит по уравнению реакции:
С6Н12О6(ж) ® 2С2Н5(ОН)(ж) 2СО2(г)
DG0обр, кДж/моль -915 -174 -394
Решение:
DG0х.р. = 2(-174) 2(-394) – (-915)= -221 кДж/моль
DН0х.р. этого процесса составляет –79 кДж/моль. Следовательно, за счет энтропийного вклада способность брожения совершать полезную работу увеличивается почти втрое. Это очень важно, так как подобные процессы служат важнейшим источником энергии как у многих так называемых анаэробных организмов, так и у животных и человека при интенсивной мышечной нагрузке.
4. Функция G называется энергией, так как определяет работоспособность соответствующего процесса. А именно, DG численно рана полезной максимальной работе, которая могла бы быть совершена в ходе изобарно-изотермического процесса.
§
План
1. Гомогенные и гетерогенные химические процессы.
2. Факторы, влияющие на скорость химических реакций.
2.1 природа реагирующих веществ;
2.2 концентрация (давление). Закон действия масс.
2.3 температура. Правило Вант-Гоффа. Уравнение Аррениуса.
Энергия активации.
2.4 катализатор.
3. Химическое равновесие. Константа равновесия.
4. Смещение равновесия. Принцип Ле-Шателье.
Литература:
1. Фролов В.В. Химия. Гл.V, §51-56.
2. Лучинский Г.П. Курс химии. Гл.V, §8-12, гл. VI, §13-18
3. Ахметов Н.С. Общая и неорганическая химия. Раздел V, гл.3,4.
4. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
Химическая термодинамика изучает возможность, направление и пределы самопроизвольного протекания химических процессов. Однако механизм и скорость процессов в химической термодинамике не рассматриваются. В то же время представление о скорости химических реакций и факторах, влияющих на нее, исключительно важно для управления химическими процессами. Область химии, изучающая механизм химических реакций и скорость их протекания, называется химической кинетикой.
Химические реакции могут протекать с различными скоростями. Одни из них протекают мгновенно: 2H2 O2 2H2O , другие протекают в течение нескольких секунд, минут:CuSO4 2NaOH Na2SO4 Cu(OH)2, FeCl3 3NaOH 3NaCl Fe(OH)3 .
2H2O , другие протекают в течение нескольких секунд, минут:CuSO4 2NaOH Na2SO4 Cu(OH)2, FeCl3 3NaOH 3NaCl Fe(OH)3 .
Биологические реакции могут длиться годы, десятилетия, а превращение древесины в уголь происходит в течение столетий, тысячелетий. При изучении скорости химических реакций необходимо различать гомогенные и гетерогенные реакции.
Гомогенные реакции– это такие реакции, в которых исходные вещества и продукты находятся в одинаковых агрегатных состояниях: газообразном (г) или жидком (ж), или твердом (т) и не имеют между собой поверхности раздела.
Гетерогенные реакции – это такие реакции, в которых исходные вещества и продукты реакции имеют между собой поверхность раздела и находятся в различных агрегатных состояниях.
В химической кинетике различают мгновенную (или истинную) скорость и среднюю скорость.
Мгновенная скорость химической реакции определяется первой производной концентрации по времени.
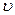 мгн
мгн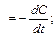 С — концентрация реагирующих веществ (моль/л);
С — концентрация реагирующих веществ (моль/л);
t –время реакции, с.
Средняя скорость реакции 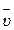 равна изменению концентрации (DС) реагирующих веществ за определенный промежуток времени (Dt):
равна изменению концентрации (DС) реагирующих веществ за определенный промежуток времени (Dt):
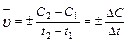
Размерность скорости реакции моль/л .с.
Скорость гомогенной реакции определяется как количество вещества (n) вступившего в реакцию или образовавшегося в результате химической реакции за единицу времени в единице объема:
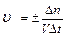 ; n – число молей участвующих в реакции;
; n – число молей участвующих в реакции;
V – объем (л);
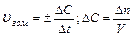 — молярная концентрация (моль/л).
— молярная концентрация (моль/л).
Скорость гетерогенной реакции – это количество вещества вступившего в реакцию или образовавшегося в результате ее за единицу времени на единице площади поверхности фазы:
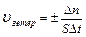 ;
; 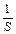 — единица площади поверхности фазы.
— единица площади поверхности фазы.
Факторы, влияющие на скорость химической реакции:
§
Говорить о скорости химической реакции имеет смысл, если природа реагирующих веществ позволяет им вступать в химическое взаимодействие. Например, природа реагирующих веществ позволяет протекать первой и второй реакции, но не позволяет третьей.
1. Mg O2 2MgО
2. Zn 2HCl ZnCl2 H2↑
3. NaOH KOH ≠
2. Зависимость скорости химической реакции от концентрацииреагирующих веществ определяется законом действия масс, который был установлен в 1867г опытным путем Гульдбергом и Вааге.
Скорость химической реакции при данной температуре пропорциональна произведению концентраций реагирующих веществ в степени, равной стехиометрическому коэффициенту, стоящему перед формулой данного вещества в уравнении реакции.
Закон действия масс справедлив только для наиболее простых по своему механизму реакций взаимодействия, протекающих в газах или в разбавленных растворах.
Для гомогенных реакций:
1.aA(Ж) bB(Ж) ↔ cC(Ж) dD(Ж) ; 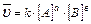 (T=const)
(T=const)
2. 3H2(Г) N2(Г) ↔ 2NH3(Г); 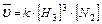
Для гетерогенных реакций:
1. aA(т) bB(Г) = cC(Г) dD(Г); 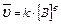 2. С (т) О2(Г)=СО2(Г);
2. С (т) О2(Г)=СО2(Г); 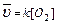
В законе действия масс не учитываются концентрации веществ, находящихся в твердой фазе. Чем больше площадь поверхности твердой фазы, тем выше скорость химической реакции.
k – константа скорости химической реакции определяется природой реагирующих веществ и зависит от температуры, от присутствия в системе катализатора, но не зависит от концентрации реагирующих веществ. Константа скорости представляет собой скорость химической реакции ( 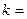
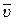 ), если концентрации реагирующих веществ
), если концентрации реагирующих веществ 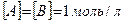 .
.
3. Зависимость скорости химической реакции от давления. Для газообразных систем увеличение давления или уменьшение объема, равноценно увеличению концентрации и наоборот.
Задача:Как изменится скорость химической реакции 2SO2(г) O2(г) 2SO3(г), если давление в системе увеличить в 4 раза?
В соответствие с законом действия масс для прямой реакции, записываем выражение:
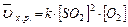 , пусть [SO2]=a моль/л, [O2]=b моль/л, тогда по закону действия масс
, пусть [SO2]=a моль/л, [O2]=b моль/л, тогда по закону действия масс 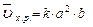
Уменьшение объема в 4 раза соответствует увеличению концентрации в системе в 4 раза, тогда
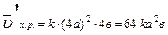 ;
;
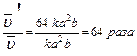 .
.
§
Влияние температуры на скорость химической реакции приближенно определяется правилом Вант-Гоффа. При повышении температуры на 100С скорость химической реакции возрастает в 2-4раза.
Математическая запись правила Вант-Гоффа: 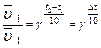 γ — температурный коэффициент скорости реакции или коэффициент Вант-Гоффадля большинства реакцийлежит в пределах 2-4.
γ — температурный коэффициент скорости реакции или коэффициент Вант-Гоффадля большинства реакцийлежит в пределах 2-4.
Задача. Во сколько раз изменится скорость химической реакции, протекающей в газовой фазе, если температура изменилась от 80 0С до 120 0С (γ = 3)?
В соответствии с правилом Вант-Гоффа записываем:
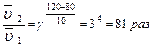
Увеличение скорости химической реакции при повышении температуры объясняется не только увеличением кинетической энергии взаимодействующих молекул. Например, число столкновений молекул растет пропорционально корню квадратному из абсолютной температуры 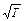 . При нагревании веществ от нуля до ста градусов по Цельсию, скорость движения молекул возрастает в 1,2 раза
. При нагревании веществ от нуля до ста градусов по Цельсию, скорость движения молекул возрастает в 1,2 раза 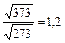 , а скорость химической реакции возрастает примерно в 59 тысяч раз. Такое резкое увеличение скорости реакции с ростом температуры объясняется долей активных молекул, столкновения которых приводит к химическому взаимодействию. Согласно теории активных столкновений в реакцию вступают только активные молекулы, энергия которых превышает среднюю энергию молекул данного вещества, т.е. молекулы, обладающие энергией активации.
, а скорость химической реакции возрастает примерно в 59 тысяч раз. Такое резкое увеличение скорости реакции с ростом температуры объясняется долей активных молекул, столкновения которых приводит к химическому взаимодействию. Согласно теории активных столкновений в реакцию вступают только активные молекулы, энергия которых превышает среднюю энергию молекул данного вещества, т.е. молекулы, обладающие энергией активации.
Энергия активации (EА)– это тот избыток энергии по сравнению со средним запасом, которым должны обладать молекулы для осуществления химической реакции. Если ЕА< 40 кДж/моль – реакции протекают быстро, если ЕА > 120 кДж/моль – реакции не идут, если ЕА = 40-120 кДж/моль – реакции протекают в обычных условиях. Повышение температуры снижает энергию активации, делает вещества более реакционно-способными, скорость взаимодействия при этом увеличивается.
Более точную зависимость скорости химической реакции от температуры установил C.Аррениус: константа скорости реакции пропорциональна основанию натурального логарифма, возведенного в степень ( –ЕА/RT). 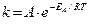 ,
,
А – предэкспоненциальный множитель, определяет число активных
соударений;
е – экспонента (основание натурального логарифма).
Логарифмируя выражение , получим уравнение:
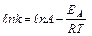 .Уравнение Аррениуса показывает, что скорость реакции тем выше, чем меньше энергия активации. Для снижения энергии активации используют катализаторы.
.Уравнение Аррениуса показывает, что скорость реакции тем выше, чем меньше энергия активации. Для снижения энергии активации используют катализаторы.
§
Катализатор – это вещество, изменяющее скорость химической реакции, количество которого в результате реакции остается неизменным. Изменение скорости химической реакции в присутствии катализаторов называется катализом. Реакции, скорость которых можно изменить при помощи катализаторов, называются каталитическими.Катализаторы, замедляющие скорость химической реакции, называются ингибиторами. Например, тетраэтилсвинец Pb(C2H5)4 – противодействует детонации топлива в двигателях внутреннего сгорания. Вещества, усиливающие действие катализатора называются промоторами. Вещества, подавляющие действие катализатора называются каталитическими ядами; биологические катализаторы называются ферментами.
Механизм катализа может быть различным. По теории промежуточных соединений катализатор (К) с одним из реагирующих веществ (А) образует промежуточное соединение АК ( активированный комплекс ) с более низким значением энергии активации, способное к более эффективному взаимодействию с другим веществом (В).
А В  АВ
АВ
А К  АК
АК
АК В  АВ К
АВ К
Таким образом, катализатор разбивает реакцию на промежуточные стадии; при этом образуется ряд неустойчивых промежуточных соединений – активированных комплексов. Например, самопроизвольный процесс разложения пероксида водорода идет медленно: Н2О2 Н2О 1/2О2 Еа = 75 кДж/моль .Использование катализатора MnO2 увеличивает скорость реакции (Еа = 49 кДж/моль ), за счет образования активированного комплекса MnO3 :
Н2О2 MnO2 H2O MnO3
 MnO3 MnO2 1/2O2 H2O2 MnO2 H2O MnO2 1/2O2
MnO3 MnO2 1/2O2 H2O2 MnO2 H2O MnO2 1/2O2
Катализатор снижает энергию активации, разбивая реакцию на ряд промежуточных стадий.
Катализатор может быть специфичен для однотипных реакций, например, V2O5 ускоряет реакции окисления SO2, NH3 и т.д. Он может быть и универсален, т.е. может изменять скорости разных реакций, например,
2SO2 O2 2SO3; CH2 = CH2 H2 CH3 – CH3. Катализатор может изменять не только скорость химической реакции, но и ее механизм.
2SO3; CH2 = CH2 H2 CH3 – CH3. Катализатор может изменять не только скорость химической реакции, но и ее механизм.
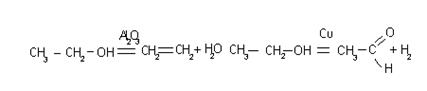
§
Для гомогенных реакций:
aA(г) bB(г) Û cC(г) dD(г), 3Н2 N2Û2NH3
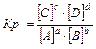 ,
, 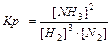
Для гетерогенных реакций:
aA(т) bB(Г) = cC(Г) dD(Г) , C(т) O2(Г) = СО2(Г)
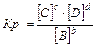
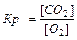
Константа равновесия зависит от природы веществ, температуры. Присутствие катализатора в системе не влияет на значение КР, поскольку он снижает ЕА прямой и обратной реакций на одну и ту же величину и поэтому одинаково изменяет скорости прямой и обратной реакций. Катализатор лишь ускоряет достижение равновесия, он не влияет на количественный выход продуктов реакции. КР не зависит от концентрации реагирующих веществ.
Химическое равновесие является подвижным (динамическим), его можно смещать, изменяя различные факторы (С; Р; T) . Сдвиг химического равновесия определяется принципом Ле-Шателье:
если на систему, находящуюся в равновесии оказывать внешнее воздействие, то система противодействует этому воздействию, стараясь его уменьшить.
Задача.
В каком направлении произойдет смещение химического равновесия системы: N2(г) 3H2(г)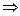 2NH3(г), ΔΗ‹ 0 – (реакция экзотермическая).
2NH3(г), ΔΗ‹ 0 – (реакция экзотермическая).
Реакция идет в прямом Реакция идет в обратном
направлении направлении


если понизить температуру t0С, еслиповысить t0C
увеличить давлние P уменьшить Р
увеличить концентрации  увеличить
увеличить 
уменьшать 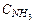 т.е.отводить NH3 ,
т.е.отводить NH3 ,
Задача. В гомогенной системе СО Сl2 COCl2 , равновесные концентрации 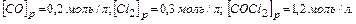 Вычислите Кр и исходные концентрации.
Вычислите Кр и исходные концентрации.
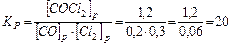 .
.
В результате реакции образовалось 1,2 моля СОСl2, учитывая коэффициенты в уравнении реакции, израсходовалось 1,2 моль/л СО и 1,2 моль/л Сl2, а так как к моменту равновесия осталось 0,2 моль/л СО и 0,3 моль/л Сl2, то

Рассматривая законы химической термодинамики, было установлено, что возможно протекание тех процессов, у которых ΔG0298<0 (Р, Т = const), а т.к. при химическом равновесии идут обратимые процессы, то какой именно процесс будет преобладать, можно определить по изменению энергии Гиббса. Мы знаем, что равновесие зависит не только от концентрации, но и от температуры и давления (для газообразных систем).
Зависимость Кр. от температуры при постоянном давлении связана с изменением ΔG0298 уравнением:
ΔG0298 = -2,3 RT lg Kр, если R = 8,31·10-3 кДж/моль·К; T = 298 K ΔG0298 = -5,7 lg Kр, кДж/моль.
Если lgKр > 0, т.е. Кр > 1, тогда ΔG0298 < 0 равновесие смещается в сторону прямой реакции.
Если lgKр < 0, т.е. Кр < 1, тогда ΔG0298 > 0 равновесие смещается в сторону обратной реакции.
§
»
План
1.1. Определение понятия «электролиты»
2.2. Общие представления теории электролитической диссоциации
а) степень диссоциации (a)Кд
б) константа диссоциации (aКд)
в) закон Оствальда
3.3. Электролитическая диссоциация воды
а) Kw – ионное произведение воды (Kw ))
б) водородный показатель (рН)
в) индикаторы
4.4. Буферные растворы
5.5. Гидролиз
Литература
1.1. Глинка Н.Л. Общая химия: Учебное пособие для вузов. – 24-е изд., исправленное/ Под ред. В.А. Рабиновича. – Л.: Химия, 1985. – с. 225-249.
2.2. Общая химия. Под ред. Е.М. Соколовской, Г.Д. Вовченко, Л.С. Гузия. М., Изд-во Московского университета, 1980. – с. 239-247, с. 262-268.
3. Лучинский Г.П. Курс химии: Учебник для инженерно-технических (нехимических) вузов. – М.: Высшая школа, 1985. – с. 169-174.
Электролиты – это вещества, которые в растворе или расплаве распадаются на ионы.
Ионы – электрически заряженные частицы, способные к самостоятельному существованию в этих средах.
Электролитическая диссоциация – распад электролита на ионы под действием молекул растворителя.
Для количественной характеристики электролитической диссоциации введено понятие степени диссоциации ( 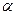 ).
).
Степень диссоциации ( ) – это отношение числа (или концентрации С) молекул распавшихся на ионы, к общему числу (или к концентрации Сисх) молекул растворенного электролита.
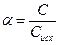
Электролиты (по заряду иона)
Таблица№12
| Бинарные или симметричные (распадаются на 2 иона) | Несимметричные |
| 1,1 валентные: NaCl à Na Cl— KBr à K Br— | 1,2 валентные: K2SO4 à 2K SO42- |
| 2,2 валентные: MgSO4 à Mg2 SO42- ZnSO4 à Zn2 SO42- | 2,1 валентные: MgCl2 à Mg2 2Cl— |
Электролиты (по степени диссоциации)
Таблица№13
Рассмотрим более подробно разбавленные растворы слабых электролитов. При изучении растворов слабых электролитов (кислот, оснований, солей) было обнаружено некоторое отклонение от законов Вант-Гоффа и Рауля, полученных для растворов неэлектролитов. Для растворов электролитов отмечались более высокие значения осмотического давления, повышение температуры кипения и понижение температуры замерзания по сравнению с растворами неэлектролитов.
Раствор слабого электролита в результате неполной диссоциации представляет собой смесь молекул электролита, ионов электролита, молекул растворителя, между которыми отсутствует взаимодействие.
Таким образом, отступление от законов Рауля и Вант-Гоффа обусловлено тем, что количество частиц в растворе электролита вследствие неполной диссоциации больше, чем в растворе неэлектролита. Вант Гофф для растворов электролитов ввел поправочный коэффициент 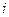 (для неэлектролитов =1, для электролитов >1), который назвал изотоническим. Уравнения для электролитов выглядят следующим образом:
(для неэлектролитов =1, для электролитов >1), который назвал изотоническим. Уравнения для электролитов выглядят следующим образом:
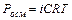
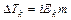
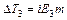
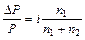
Взаимосвязь изотонического коэффициента ( ) со степенью диссоциации ( 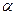 ) можно найти, используя следующие рассуждения.
) можно найти, используя следующие рассуждения.
Рассмотрим случай, когда молекула электролита распадается на 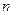 ионов (
ионов ( 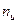 ) со степенью диссоциации . Тогда количество ионов составляет:
) со степенью диссоциации . Тогда количество ионов составляет: 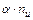 , а количество недиссоциированных молекул:
, а количество недиссоциированных молекул: 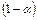 .
.
Среднее число частиц, образующихся из одной молекулы можно записать:
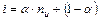 ,
,
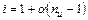 ,
,
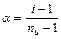 .
.
Последнее уравнение позволяет вычислить степень диссоциации слабого электролита, если изотонический коэффициент определен экспериментально (например, по изменению температуры замерзания раствора).
Итак, слабый
электролит в растворе диссоциирует на ионы, например, уксусная кислота: СН3СООН à СН3СОО— Н
Процесс, обратный диссоциации – рекомбинация ионов в молекулу:
СН3СООН ß СН3СОО— Н
С течением времени устанавливается динамическое равновесие между молекулярной и ионной формами электролита в растворе:
СН3СООН « СН3СОО— Н
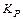 характеризует это состояние:
характеризует это состояние: 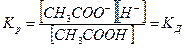
Константа диссоциации 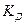 – это константа равновесия (
– это константа равновесия ( 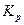 ), отвечающая диссоциации слабого электролита.
), отвечающая диссоциации слабого электролита.
зависит от: 1.Температуры
2. 2.Природы веществ, растворителя
2.
не зависит от:
1.Концентрации.
Ступенчатая диссоциация характерна, для слабых многоосновных кислот (например, Н2СО3):
a) Н2СО3 « Н НСО3—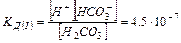 I ступень
I ступень
b) НСО3— « Н СО32-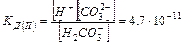 II ступень
II ступень
________________________________________________________________________________________________
Н2СО3 « 2Н СО32-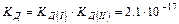
Ступенчатая диссоциация для слабых многокислотных оснований:
Fe(OH)3 « [Fe(OH)2] OH— KД(I) I ступень
[Fe(OH)2] « [FeOH]2 OH— KД(II) II ступень [FeOH]2 « Fe3 OH— KД(III) III ступень
______________________________________________
Fe(OH)3 « Fe3 3OH— KД = КД(I) КД(II) КД(II
КД(I) > КД(II) > КД(III) (распад происходит в меньшей степени по последующим ступеням).
§
Важной особенностью жидкой воды является ее способность к самопроизвольной диссоциации по реакции:
Н2О(ж) « Н (водн) ОН—(водн)
Этот процесс называется еще самоионизацией или автопротолизом. Образовавшиеся протоны Н и анионы ОН— окружены определенным числом полярных молекул воды, т.е. гидратированы: Н ×nH2O; OH—×mH2O. Первичная гидратация может быть представлена рядом аквакомплексов: Н3О ; Н5О2 ; Н7О3 ; Н9О4 , среди которых преобладают ионы Н9О4 (Н ×4H2O). Время жизни всех этих ионов в воде очень мало, т.к. протоны постоянно мигрируют от одних молекул
воды к другим. Обычно в уравнениях для простоты используют только катион состава Н3О (Н ×H2O), называемый ионом гидроксония.
Процесс диссоциации воды с учетом гидратации протона и образования иона гидроксония может быть записан: 2Н2О « Н3О ОН—
Вода – слабый электролит, степень диссоциации которого
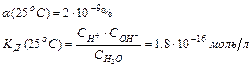
Поскольку 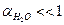 à Сравн(Н2О)» Сисх(Н2О) или [Н2О]равн ≈ [Н2О]исх
à Сравн(Н2О)» Сисх(Н2О) или [Н2О]равн ≈ [Н2О]исх
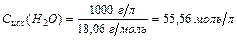 – количество молей содержащееся в одном литре воды. Сисх(Н2О) в разбавленном растворе остается постоянной. Это обстоятельство позволяет включить Сравн(Н2О) в константу равновесия.
– количество молей содержащееся в одном литре воды. Сисх(Н2О) в разбавленном растворе остается постоянной. Это обстоятельство позволяет включить Сравн(Н2О) в константу равновесия.
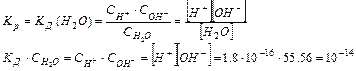
Таким образом, произведение двух постоянных величин 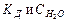 дает новую постоянную, которую называют ионным произведением воды
дает новую постоянную, которую называют ионным произведением воды 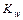 . При температуре 298 К
. При температуре 298 К  .
.
¾- Постоянство ионного произведения воды означает, что в любом водном растворе: кислотном, нейтральном или щелочном – всегда имеются оба вида ионов (Н и ОН—)
¾- В чистой воде концентрации водородных и гидроксильных ионов равны и при нормальных условиях составляют:
[H ] = [OH—] = Kw1/2 = 10-7 моль/л.
¾- При добавлении кислот концентрация [Н ] увеличивается, т.е. равновесие смещается влево, а концентрация [ОН—] уменьшается, однако Кw остается равной 10-14.
В кислой среде [H ] > 10-7 моль/л, а [OH—] < 10-7 моль/л
В щелочной среде [H ] < 10-7 моль/л, а [OH—] > 10-7 моль/л
На практике для удобства используют водородный показатель (рН) и гидроксильный показатель (рОН) среды.
Это есть взятый с обратным знаком десятичный логарифм концентраций (активностей) соответственно ионов водорода или гидроксильных ионов в растворе: рН = — lg[H ], рОН = — lg[OH—]
В водных растворах рН рОН = 14.
Таблица№14.
| Растворы | ||
| Нейтральные | Кислые | Щелочные |
| рН = 7 | рН < 7 | рН > 7 |
Kw зависит от температуры (т.к. диссоциация воды – эндотермический процесс)
Kw (25 oC) = 10-14 Þ рН = 7
Kw (50 oC) = 5,47×10-14 Þ рН = 6,63
Измерение рН используется чрезвычайно широко. В биологии и медицине величина рН биологических жидкостей служит для определения патологий. Например, в норме рН сыворотки крови состовляет 7,4±0,05; слюны – 6,35..6,85; желудочного сока – 0,9..1,1; слез – 7,4±0,1. В сельском хозяйстве рН характеризует кислотность почв, экологическое состояние природных вод и т.д.
Кислотно-основными индикаторами называются химические соединения, изменяющие свою окраску в зависимости от рН среды, в которой они находятся. Вы, наверное, обращали внимание на то, как меняется цвет чая, если в него положить лимон – это пример действия кислотно-основного индикатора.
Индикаторы, как правило, представляют собой слабые органические кислоты или основания и могут существовать в растворах в виде двух таутомерных форм:
HInd « H Ind—, где HInd – кислотная форма (это форма, которая преобладает в кислых растворах); Ind – основная форма (преобладает в щелочных растворах).
Поведение индикатора подобно поведению слабого электролита в присутствии более сильного с одноименным ионом. Чем больше [H ] следовательно равновесие смещается в сторону существования кислотной формы HInd и наоборот (принцип Ле-Шателье).
Опыт показывает наглядно возможность использования некоторых индикаторов:
Таблица№15
| Индикатор | рН < 7 | рН = 7 | рН > 7 |
| Метилоранж | красный | оранжевый | желтый |
| Фенолфталеин | бесцветный | бесцветный | малиновый |
| Лакмус | красный | Фиолетовый | синий |
Специальные приборы – рН-метры позволяют измерять рН с точностью до 0,01 в интервале от 0 до 14. Определение основано на измерении ЭДС гальванического элемента, один из электродов которого является, например, стеклянным.
Наиболее точно концентрацию водородных ионов можно определить методом кислотно-основного титрования. Титрование – это процесс постепенного добавления небольшими порциями раствора известной концентрации (титранта) к титрируемому раствору, концентрацию которого хотим определить.
Буферные растворы – это системы, рН которых относительно мало изменяется при разбавлении или добавлении к ним небольших количеств кислот или щелочей. Чаще всего они представляют собой растворы, содержащие:
a) а)Слабую кислоту и ее соль(СН3СООН СН3СООNa) – ацетатный буфер
в)Слабое основание и его соль(NH4OH NH4Cl) – аммиачно-аммонийный буфер
с)Две кислые соли с разными Kд (Na2HPO4 NaH2PO4) – фосфатный буфер
Регулирующий механизм буферных растворов рассмотрим на примере ацетатного буферного раствора.
CH3COOH « CH3COO— H , 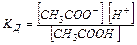
CH3COONa « CH3COO— Na
1. 1)если добавить небольшое количество щелочи к буферной смеси:
CH3COOH NaOH « CH3COONa H2O,
NaOH нейтрализуется уксусной кислотой с образованием более слабого электролита H2O. Избыток натрия ацетата смещает равновесие в сторону образовавшейся кислоты.
2. 2)если добавить небольшое количество кислоты:
CH3COONa HCl « CH3COOH NaCl
Катионы водорода Н связывают ионы CH3COO—
Найдем концентрацию ионов водорода в буферном ацетатном растворе:
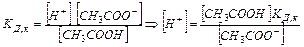
Равновесная концентрация уксусной кислоты рана Cисх,к (т.к. слабый электролит), а [СH3COO—] = Cсоли (т.к. соль является сильным электролитом), то 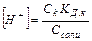 . Уравнение Гендерсона-Хассельбаха:
. Уравнение Гендерсона-Хассельбаха:
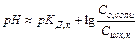
Таким образом, рН буферных систем определяется соотношением концентраций соли и кислоты. При разбавлении это соотношение не меняется и рН буфера не меняется при разбавлении, это отличает буферные системы от раствора чистого электролита, для которого справедлив закон разведения Оствальда.
Существует две характеристики буферных систем:
1.Буферная сила. Абсолютная величина буферной силы зависит от
общей концентрации компонентов буферной системы, т.е. чем больше концентрация буферной системы, тем больше требуется щелочи (кислоты) для одного и того же изменения рН.
2.Буферная емкость (В). Буферная емкость – это предел, в котором проявляется буферное действие. Буферная смесь поддерживает рН постоянным только при условии, что количество прибавляемых к раствору сильной кислоты или основания не превышает определенной предельной величины – В. Буферная емкость определяется числом г/экв сильной кислоты (основания), которое необходимо прибавить к одному литру буферной смеси, чтобы изменить значение рН на единицу, т.е.  . Вывод: Свойства буферных систем:
. Вывод: Свойства буферных систем:
1. 1.[H ] мало зависит от разбавления.
2. 2.Прибавление сильных кислот (оснований) мало изменяет [H ] в пределах буферной емкости В.
3. 3.Буферная емкость зависит от буферной силы (от концентрации компонентов).
4. 4.Максимальное действие проявляет буфер в случае, когда кислота и соль присутствуют в растворе в эквивалентных количествах:
Ссоли = Ск-ты; [H ] = Кд,к; рН = рКд,к (рН определяется значением Кд).
Гидролиз – это химическое взаимодействие воды с солями. Гидролиз солей сводиться к процессу передачи протонов. В результате его протекания появляется некоторый избыток водородных или гидроксильных ионов, сообщающих раствору кислотные или щелочные свойства. Таким образом, гидролиз обратен процессу нейтрализации.
Гидролиз солей включает 2 стадии:
а)Электролитическая диссоциация соли с образованием гидратированных ионов:. KCl à K Cl— K xH2O à K × xH2O (связь донорно-акцепторная, донор – атом О, имеющий 2 неподеленные электронные пары,
акцептор – катионы с вакантными орбиталями)
Cl— yH2O « Cl— ×yH2O (водородная связь)
в) Гидролиз по аниону. Cl— HOH à HCl OH—
с) Гидролиз по катиону. K HOH à KOH
Гидролизу подвергаются все соли, образованные с участием слабых
электролитов:
1.Соль, образованная анионом слабых кислот и катионом сильных оснований [CH3COONa, NaClO, KNO2, Na2CO3, Na3PO4]
CH3COONa HOH « CH3COOH NaOH
CH3COO— НОН « CH3COOН OH—, рН > 7
Анионы слабых кислот выполняют функцию оснований по отношению к воде – донору протонов, что приводит к увеличению концентрации ОН—, т.е. подщелачиванию среды.
Глубина протекания гидролиза определяется: степенью гидролиза aг:
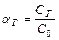 ,
,
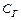 – концентрация соли, подвергшейся гидролизу
– концентрация соли, подвергшейся гидролизу
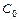 – концентрация исходной соли
– концентрация исходной соли
aг невелика, например, для 0,1 моля раствора CH3COONa при 298 К она равна 10-4.
При гидролизе в системе устанавливается равновесие, характеризующееся Кр
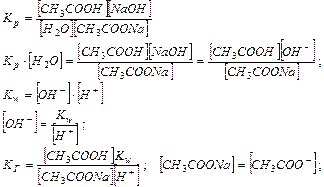
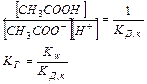
Следовательно, чем меньше константа диссоциации, тем больше константа гидролиза. Степень гидролиза с константой гидролиза связана уравнением:
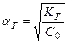 .
.
С увеличением разбавления, т.е. уменьшением С0, степень гидролиза увеличивается.
2. 2.Соль, образованная катионом слабых оснований и анионом сильных кислот [NH4Cl, AgNO3, ZnCl2, Fe2(SO4)3]
NH4Cl HOH ↔ NH4OH
HCl
NH4 HOH ↔ NH4OH H , pH < 7
Протолитическое равновесие смещено влево, катион слабого основания NH4 выполняет функцию кислоты по отношению к воде, что приводит к подкислению среды. Константа гидролиза определяется по уравнению:
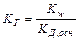
Равновесная концентрация ионов водорода может быть вычислена: [Н ]равн = aг × С0 (исходная концентрация соли), где
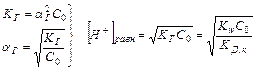 ;
;
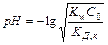
Кислотность среды зависит от исходной концентрации солей подобного вида.
3. 3.Соль, образованная анионом слабых кислот и катионом слабых оснований. Гидролизуется и по катиону и по аниону [NH4CN, CH3COONH4, (NH4)2S, (NH4)2CO3, (NH4)3PO4, ZnS, AlPO4, Al(CH3COO)3, C4(HCOO)2]
NH4CN HOH à NH4OH HCN
Для определения рН среды раствора сравнивают КД,к и КД,осн
КД,к > КД,осн à среда слабо кислая
КД,к < КД,осн à среда слабо щелочная
КД,к = КД,осн à среда нейтральная
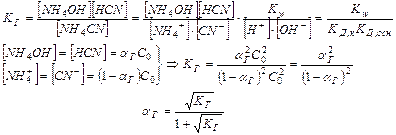
Следовательно, степень гидролиза этого вида солей не зависит от их концентрации в растворе.
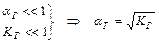
т.к. [H ] и [ОН—] определяются КД,к и КД,осн, то 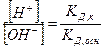
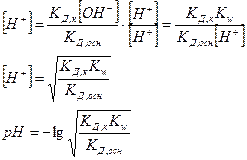
рН раствора также не зависит от концентраций соли в растворе.
Соли, образованные многозарядным анионом и однозарядным катионом (сульфиды, карбонаты, фосфаты аммония) практически полностью гидролизуются по первой ступени, т.е. находятся в растворе в виде смеси слабого основания NH4OH и его соли NH4HS, т.е. в виде аммонийного буфера.
Для солей, образованных многозарядным катионом и однозарядным анионом (ацетаты, формиаты Al, Mg, Fe, Cu) гидролиз усиливается при нагревании и приводит к образованию основных солей.
Гидролиз нитратов, гипохлоритов, гипобромитов Al, Mg, Fe, Cu протекает полностью и необратимо, т.е. соли не выделены из растворов.
Соли: ZnS, AlPO4, FeCO3 и др. в воде малорастворимы, тем не менее часть их ионов принимает участие в процессе гидролиза, это приводит к некоторому возрастанию их растворимости.
Сульфиды хрома и алюминия гидролизуются полностью и необратимо с образованием соответствующих гидроксидов.
4. 4.Соли, образованные анионом сильных кислот и сильных оснований гидролизу не подвергаются [KCl, Na2SO4, Ba(NO3)2].
Чаще всего гидролиз ‑ вредное явление, вызывающее различные осложнения. Так при синтезе неорганических веществ из водных растворов в получаемом веществе появляются примеси – продукты его гидролиза. Некоторые соединения из-за необратимо протекающего гидролиза вообще не удается синтезировать.
·-если гидролиз протекает по аниону, то в раствор добавляют избыток щелочи
·-если гидролиз протекает по катиону, то в раствор добавляют избыток кислоты
Итак, первая качественная теория растворов электролитов была высказана Аррениусом (1883 – 1887 г.). По этой теории:
1. 1.Молекулы электролита диссоциируют на противоположные ионы
2. 2.Между процессами диссоциации и рекомбинации устанавливается динамическое равновесие, которое характеризуется КД. Это равновесие подчиняется закону действия масс. Долю распавшихся молекул характеризует степень диссоциации a. КД и a связывает закон Оствальда.
3. 3.Раствор электролита (по Аррениусу) – это смесь молекул электролита, его ионов и молекул растворителя, между которыми отсутствует взаимодействие.
Вывод: теория Аррениуса позволила объяснить многие свойства растворов слабых электролитов при небольшой концентрации.
Однако, теория Аррениуса носила только физический характер, т.е. не рассматривала вопросы:
· По какой причине вещества в растворах распадаются на ионы?
· Что происходит с ионами в растворах?
Дальнейшее развитие теория Аррениуса получила в работах Оствальда, Писаржевского, Каблукова, Нернста и т.д. Например, на важное значение гидратации впервые указал Каблуков (1891), положив начало развитию теории электролитов в направлении, которое указывал Менделеев (т.е. ему впервые удалось объединить сольватную теорию Менделеева с физической теорией Аррениуса). Сольватация – это процесс взаимодействия электролита
молекулами растворителя с образованием комплексных соединений сольватов. Если растворителем является вода, следовательно, процесс взаимодействия электролита с молекулами воды называется гидратацией, а аквакомплексы – кристаллогидратами.
Рассмотрим пример диссоциации электролитов, находящихся в кристаллическом состоянии. Этот процесс возможно представить в две стадии:
1. 1.разрушение кристаллической решетки вещества DН0кр > 0, процесс образования молекул (эндотермический)
2. 2.образование сольватированных молекул, DН0сольв < 0, процесс экзотермический
Результирующая теплота растворения равна сумме теплот двух стадий DН0раств = DН0кр DН0сольв и может быть как отрицательной, так и положительной. Например, энергия кристаллической решетки KCl = 170 ккал/моль.
Теплота гидратации ионов К = 81 ккал/моль, Cl— = 84 ккал/моль, а результирующая энергия равна 165 ккал/моль.
Теплота гидратации частично покрывает энергию необходимую для выделения ионов из кристалла. Оставшиеся 170 — 165 = 5 ккал/моль могут быть покрыты за счет энергии теплового движения, и растворение сопровождается поглощением теплоты из окружающей среды. Гидраты или сольваты облегчают эндотермический процесс диссоциации, затрудняя рекомбинацию.
А вот ситуация, когда присутствует только одна из двух названных стадий:
1. 1.растворение газов – нет первой стадии разрушения кристаллической решетки, остается экзотермическая сольватация, следовательно растворение газов, как правило, экзотермично.
2.при растворении кристаллогидратов отсутствует стадия сольватации, остается лишь эндотермическое разрушение кристаллической решетки. Например, раствор кристаллогидрата: CuSO4 × 5H2O (т) à CuSO4 × 5H2O (р)
DНраств = DНкр = 11,7 кДж/моль
Раствор безводной соли: CuSO4 (т) à CuSO4 (р) à CuSO4 × 5H2O (р)
DНраств =DНсольв DНкр = — 78,2 11,7 = — 66,5 кДж/моль
§
План
1. Значение комплексных соединений (КС).
2. Координационная теория строения КС А.Вернера. Природа связи в КС.
Классификация КС.
3. Номенклатура КС.
4. Пространственное строение КС. Магнитные и оптические свойства КС.
5. Устойчивость КС.
6. Получение и разрушение КС.
Литература.
1. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
2. Курс общей химии под ред. Коровина Н.В. и др. Гл.X, §X.l-X.5.
Комплексные или координационные соединения (КС) составляют наиболее разнообразный класс неорганических веществ, имеющий большое значение в природе и технике. По своему составу КС являются более сложными, чем оксиды, гидроксиды, кислоты и соли. Реакции комплексообразования лежат в основе получения редких и драгоценных металлов, чистых и сверхчистых веществ, аналитических определений, гальванотехнических процессов, получения лекарств, антидетонаторов, антиоксидантов и т.д.
Неоценима роль КС в биологических процессах. Такие жизненно важные природные соединения, как гемоглобин, хлорофилл, инсулин, некоторые витамины относятся также к координационным соединениям.
Строение и механизм образования химических связей в КС наиболее удачно обьясняет координационная теория, предложенная в 1893 г. А.Вернером. Причиной комплексообразования может быть как электростатическое, так и донорно-акцепторное взаимодействие. Рассмотрим механизм образования молекулы [Ag(NH3)2]Cl: AgCl 2NH4OH ® [Ag (NH3) 2] Cl 2H2O
В молекуле [Ag(NH3)2]Cl ион серебра имеет следующее строение: Ag0 (4d105s1)  Ag (4d105s0), ион хлора -соответственно:
Ag (4d105s0), ион хлора -соответственно:
Cl0 (3s23p5)  Cl— (3s23p6).
Cl— (3s23p6).
Взаимодействие катиона Ag  c анионом Cl¯ носит электростатический характер: Ag Cl— AgCl ионная связь . При образовании комплексного иона [Ag(NH3)2] катион Ag выступает в качестве акцептора готовой электронной пары, предоставляя, свободные орбитали (5s и 5p) для координации; молекулы аммиака выступают в качестве донора электронной пары. Химическая связь такого вида называется донорно-акцепторной или координационной. По своей природе — это полярная ковалентная связь, отличие состоит лишь в способе её образования. Координационные соединения состоят из внешней и внутреннейсфер.
c анионом Cl¯ носит электростатический характер: Ag Cl— AgCl ионная связь . При образовании комплексного иона [Ag(NH3)2] катион Ag выступает в качестве акцептора готовой электронной пары, предоставляя, свободные орбитали (5s и 5p) для координации; молекулы аммиака выступают в качестве донора электронной пары. Химическая связь такого вида называется донорно-акцепторной или координационной. По своей природе — это полярная ковалентная связь, отличие состоит лишь в способе её образования. Координационные соединения состоят из внешней и внутреннейсфер.
[H3N:←□ Ag□←: NH3] Cl‾
внутренняя сфера внешняя сфера
Различают внутренние сферы
1.Катионного характера: [Ag(NH)2] ;[Cu(NH3)4] 
2.Анионного характера: [Co(CN)4]  ; [Fe(CN)6]
; [Fe(CN)6] 
3.Нейтрального характера: [Fe (CO) 5]
Комплексную частицу, например,  Cr3 (H2O)0 6
Cr3 (H2O)0 6  3 образуют:
3 образуют:
— Центральный атом (ЦА) или, его так же называют комплексообразователь (КО) – это катион хрома Cr3
— Лиганды (L) — в нашем примере это молекулы воды, которые координируются вокруг ЦА, их количество (6) соответствует значению координационного числа (КЧ = 6).
Заряд комплексного иона равен алгебраической сумме зарядов ЦА и L:
К2 Pt2 Cl4—2-; Cr3 (H2O)0 6 3 Cl3—
В качестве комплексообразователей может быть любой элемент периодической системы, но наибольшую склонность к комплексообразованию имеют d- и f- металлы, некоторые р-элементы.
Координационное число (КЧ) — определяет количество s- связей, образуемых центральным атомом с лигандами (табл.№ 16)
Координационное число зависит от многих факторов: RЦА, заряда ЦА, размеров и валентности лигандов, среды в которой протекает реакция комплексообразования.
Таблица №16 Зависимость КЧ от заряда ЦА
| Заряд ЦА | КЧ | Пример |
| 1 | 2,3,4, но чаще всего 2 | Ag , Cu , Au [Ag (NH3) 2] Cl |
| 2 | 3,4,5,6,7,8, но чаще всего 4 | Cu2 , Fe2 , Zn2 , Hg2 , Au2 , Pt2 , Pb2 [Cu (H2O) 4]SO4, Na2[Zn(OH)4], K4 [Fe(CN)6] |
| 3 | 4,5,6,8,9,10,12, но чаще всего 6 | Fe3 , Cr3 , Co3 , Al3 , K3 [Fe (CN) 6] |
| 4 | 6,8, | Pt4 ,Pb4 , K2 [PtCl6] , K4 [PtCl8] |
Лигандамимогут быть нейтральные молекулы или анионы, простые или сложные молекулы органических веществ, у которых имеются неподеленные электронные пары.
По числу химических связей, образуемых лигандами с центральным атомом, лиганды можно разделить на:
— монодентатные : F‾; Cl‾; I‾; 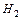
 ;
; 
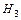
 и т.д..
и т.д..
-бидентатные — :SO4²‾ ;:NH2-(CH2)2 –Н2N: — этилендиаммин (ЭДА) .
— полидентатные: :OOCH2C CH2COО:
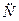 -(CH2)2—
-(CH2)2—
:OOCH2С CH2COO:
Этилендиаминтетрауксусная кислота
По наличию заряда, выделяют лиганды:
-анионного типа: CN‾; Cl‾ ; OH‾ ; CNS‾
-нейтрального типа: NH3; H2О ; CO ; NO ; ЭДА .
Комплексные соединения по химической природе лигандов делятся на четыре класса.
§
[Cr (H2O)5Cl]Cl2 [Co (NH3) 3(NO2)3]
Номенклатура комплексных соединений.
В настоящее время единый подход к решению вопроса о номенклатуре КС отсутствует. Украинские ученые предлагают:
1. в названиях КС первым указывать катион, вторым — анион.
2. в названиях комплексных ионов или молекул лиганды перечислять в алфавитном порядке независимо от заряда. Анионным лигандам приписывать окончание «о», нейтральные лиганды называть, как соответствующие молекулы, за исключением H2O-аква , NH3-аммин. Количество лигандов обозначать численными приставками : ди -; три -; тетра -; пента -; гекса — и т.д..
3.В названиях комплексных анионов комплексообразователю присваивать суффикс – «ат».
4.Степень окисления Ц.А. указывать в скобках римскими цифрами. Например:
[Co(NH3)5Cl]Cl2- пентаамминхлорокобальта (IV) хлорид
K4[Fe(CN)6]-калия гексацианоферрат (II)
[Mo6(H2O)6Cl8]Cl4- гексаакваоктахлорогексамолибдена (II) хлорид
В процессе образования КС по донорно-акцепторному механизму орбитали ЦА претерпевают гибридизацию. Тип гибридизации орбиталей комплексообразователя определяет геометрию комплексного иона. Например,
Таблица № 17
Различия в магнитных свойствах веществ могут быть объяснены электронным строением их атомов, ионов и молекул. Диамагнитными (немагнитными) считаются частицы, суммарный магнитный момент которых равен нулю, т.е. все электроны спарены и их магнитные моменты взаимно компенсируются. Если частица имеет один или несколько неспаренных электронов, то суммарный магнитный момент отличен от нуля, такая частица является парамагнитной.
Свойства КС зависят также от природы L. По силе воздействия на валентные электроны ЦА лиганды располагают в спектрохимический ряд:
 CO>CN->NO2->NH3>CNS->H2O>OH->F->Cl->Br—>I-
CO>CN->NO2->NH3>CNS->H2O>OH->F->Cl->Br—>I-

L сильного поля L поля средней силы L слабого поля
В случае сильного поля лигандов образуются спин-спаренные КС (табл. 2 , соед. 3;5),а в случае слабого поля – спин-свободные КС (табл. 2, соед 4;6) Многие КС имеют окраску. Это объясняется возможностью перехода электронов с одного d-энергетического подуровня на другой при наличии незаполненных орбиталей. При поглощении веществами определенной части спектра само вещество оказывается окрашенным в дополнительный цвет. Например, почему соединения Au(I) не окрашены, а Au(III) окрашены? Ион Au имеет электронную конфигурацию 5d10. Все 5d-орбиталей заполнены, переход электронов на d-подуровень невозможен. У иона Au3 5d8-электронов, следовательно, на d-уровне две вакансии, переход электронов в пределах d-подуровня возможен, поэтому соединения Au3 окрашены.
Ионы внешней сферы связаны с комплексным ионом силами электростатического взаимодействия, поэтому в растворах КС диссоциируют подобно сильным электролитам на внутреннюю и внешнюю сферы ( первичная диссоциация). Например:
K3[Fe(CN)6] ↔ 3K [Fe(CN)6]3- добавляя к раствору комплексного соединенияK3[Fe(CN)6] роданид калияKCNS красный цвет, характерный для соединения Fe(CNS)3 не появляется.
[Аg (NH3) 2] Cl ↔ [Ag (NH3) 2] Cl—
Если КС является нейтральной частицей, то ей не присуща первичная диссоциация.
Комплексный ион диссоциирует значительно слабее (подобно слабым электролитам)– это вторичная диссоциация, она может идти ступенчато и характеризуется КД, которая называется константой нестойкости (Кнест).
[Ag(NH3)2] ↔ [Ag(NH3)] NH3I ступень, К нест 1
[Ag(NH3)] ↔ Ag NH3II ступень, К нест 2
[Ag(NH3)2] → Ag 2NH3, К нест=Кнест1. К нест2
Эта величина является мерой устойчивости комплексного иона. Чем меньше значение Кнест, тем прочнее комплексный ион. Например, из комплексных ионов [Fe(CN)6]3- (Кнест= = 1,0 . 10-44), [Fe(CN)6]4-( Кнест =1,0 . 10-37) и [Ag(NH3)2] ( Кнест= = 2,8. 10-8) — самым прочным являетя комплексный ион гексацианоферрата [Fe(CN)6]3-.
Для получения КС можно использовать реакции соединения, замещения, двойного обмена, окислительно-восстановительные реакции (ОВР).
1) реакции соединения
CuCl2 4H2O → [Cu (H2O) 4] Cl2
Cr (OH) 3 3NaOH → Na3 [Cr (OH) 6]
Cерозеленый зеленый
3KCN Fe (CN) 3 → K3 [Fe (CN) 6]
2) реакции внешнесферного обмена
3Fe2 SO4 2K3[Fe3 (CN) 6] → Fe3 [Fe (CN) 6]2↓ 3K2SO4
синий
3) реакции внутрисферного обмена
[Co (H2O) 4] Cl2 4KCNS → K2 [Co (CNS) 4] 2KCl 4H2O
розовый фиолетовый
4) окислительно-восстановительные реакции
2K4[Fe2 (CN) 6] Cl2 → 2K3[Fe3 (CN)6] 2KCl
Разрушение комплексных соединений можно осуществлять нагреванием, разбавлением, связыванием лигандов в более прочные соединения.
[Cu (NH3) 4](OH) 2 CuO H2O 4NH3↑
CuO H2O 4NH3↑
K2 [Co (CNS) 4] 4H2O → [Co (H2O) 4](CNS) 2 2KCNS
[Ag (NH3) 2] Cl 2HNO3(Kонц) → AgCl↓ 2 NH4NO3
[Cu (NH3) 4](OH) 2 Na2S → CuS↓ 4NH3↑ 2NaOH
§
План
1. Окислительно-восстановительные реакции (ОВР). Их классификация.
2. Направление протекания ОВР.
3. Основы электрохимии:
3.1. двойной электрический слой; равновесный электродный потенциал;
3.2. стандартный электродный потенциал;
3.3. электрод сравнения (стандартный водородный электрод);
3.4. уравнение Нернста;
3.5. ряд напряжений металлов.
Литература:
1. Фролов В.В. Химия. Гл.V, §51-56.
2. Лучинский Г.П. Курс химии. Гл.V, §8-12, гл. VI, §13-18
3. Ахметов Н.С.Общая и неорганическая химия. Раздел V, гл.3,4.
4. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
Электрохимия– это раздел химии, который изучает химические процессы, в результате которых возникает электрический ток или химические процессы, протекающие под действием электрического тока.
В основе таких химических процессов лежат ОВР.
Окислительно-восстановительные реакции– это электронодинамические реакции, сопровождающиеся переходом электронов от восстановителя к окислителю и изменением степеней окисления атомов, входящих в состав реагирующих веществ.
Степень окисления (С.О.) – это формальный электрический заряд атома элемента в химическом соединении, при условии, что все образуемые этим атомом связи носят чисто ионный характер.
Степень окисления элементов определяется с учетом следующих правил:
1.С.О. (элемента в простом веществе)=0
2.C.O. (O)= -2(искл.F2O 2;R2O-12).
3.С.О. (Н)= 1(искл. гидриды металлов Na H—).
4.С.О. (иона)=его заряду Z (Na  C.O.= 1; F— C.O.= -1)
C.O.= 1; F— C.O.= -1)
5.Закон электронейтральности: в молекуле алгебраическая сумма степеней окисления элементов с учетом числа их атомов равна нулю. Например, K 1MnxO-24
1 x 4•(-2)=0 x= 7
Окислитель– вещество, которое принимает электроны и при этом восстанавливается.
Важнейшие окислители:
1.Неметаллы, переходящие в процессе ОВР в отрицательно заряженные ионы:
F02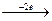 2F — (Cl2; I2; O2; Br2; S),
2F — (Cl2; I2; O2; Br2; S),
2. Катионы металлов и катион водорода Н , переходящие в процессе ОВР в нейтральное состояние:
Ag 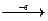 Ag0
Ag0
2H 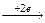 H02
H02
3. Сложные вещества, в состав которых входят атомы элементов, в высших степенях окисления, переходящие в процессе ОВР в более низкие степени окисления.
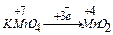
Восстановитель– это вещество, которое отдает электроны и при этом окисляется.
Важнейшие восстановители:
1.Все металлы и некоторые неметаллы(H2; B; C) переходящие в процессе ОВР в состояние с положительными степенями окисления.
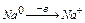
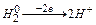
2.Сложные вещества, в состав которых входят элементы с низкими степенями окисления, переходящие при ОВР в более высокие степени окисления.
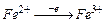
(FeCl2) (FeCl3)
Типы ОВР:
1.Межмолекулярные ОВР – реакции, в ходе которых окислитель и восстановитель находятся в разных веществах:
Mn 4O2 4HCl—=Mn2 Cl2 Cl02 2H2O
2.Внутримолекулярные ОВР – реакции, в ходе которых окислитель и восстановитель (атомы разных элементов) находятся в составе одного вещества: 2KCl 5O-23=2KCl-1 3O02.
3.ОВР диспропорционирования – реакции, в ходе которых атомы одного и того же элемента, находящиеся в составе вещества, являются и окислителем и восстановителем:
Cl02 2KOH=KCl— KCl 1O H2O
При составлении уравнений ОВР должен соблюдаться электронный баланс, т.е. число электронов, отданных восстановителем, должно соответствовать числу электронов, принятых окислителем. Вычислив степени окисления участвующих и образующихся в результате реакции частиц, определяем восстановитель и окислитель.

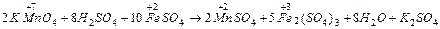
окислитель [Mn 7O4]— 8H 5ē → Mn2 4H2O 5 2 Е0= 1,51 (В)
восстановитель Fe2 — ē → Fe3 1 10 ЕВ= 0,77(В)
 Для написания молекулярного уравнения дописываем противоионы.
Для написания молекулярного уравнения дописываем противоионы.
2[MnO]—4 16H 10Fe2 → 2Mn2 10Fe3 8H2O
2K 8[SO4]2- 10SO42- →2[SO4]2- 15[SO4]-2 2K SO4 
В растворах стремление электронов к переходу от одних атомов или ионов к другим характеризуется стандартным окислительно-восстановитель-ным потенциалом Е0(В).
Возможность и направление протекания ОВР определяется величиной ЭДС, которая расчитывается как разность потенциалов окислителя (Е0) и восстановителя (ЕВ).
ЭДС = Е0 – ЕВ, если ЭДС = ΔΕ>0 реакция возможна;
ΔΕ >0,2 (В) идет хорошо; ΔΕ <0 реакция невозможна.
Для приведенной реакции ЭДС=Е0-ЕВ=1,51-0,77=0,74(В), значит, реакция протекает в прямом направлении достаточно хорошо.
§
При погружении металлической пластинки, свободной от оксидной пленки, в раствор электролита на границе металл — раствор электролита возникает двойной электрический слой (ДЭС), характеризующийся определённой разностью потенциалов или определённым электродным потенциалом.
ДЭС возникает в результате перехода заряженных частиц через границу раздела двух фаз металл (М), твердая фаза (т) — раствор электролита (ж). При его формировании, например, в случае металлической пластинки, погруженной в воду, протекают следующие процессы:
1. Отрыв положительно заряженных ионов (катионов) от поверхности металла и переход их в раствор.
М (т)→М 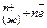
2. Избыток электронов, остающихся в металле, заряжает его поверхность отрицательно.
3. Гидратация катионов полярными молекулами воды, в объёме жидкой фазы (сольватация молекулами растворителя)
М  nH
nH  O→М
O→М  ·nH O
·nH O 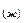
Выше перечисленные процессы (1,2,3)протекают одновременно и поэтому их общее уравнение можно записать:
М (т) nH O→М ·nH O n 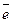
4. Концентрирование гидратированных (сольватированных) катионов в прилегающем к поверхности металла слое жидкости вследствие их взаимодействия с отрицательно заряженной поверхностью. Положительно заряженные катионы притягиваются отрицательно заряженной поверхностью. Формируется ДЭС, характеризующийся электродным потенциалом.
5. С ростом скачка потенциала между металлом и раствором электролита всё с большей скоростью начинают протекать обратные процессы, дегидратация ионов металла и их последующее восстановление до атомов.
М (ж))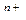 ·nH O n → М (т) nH O
·nH O n → М (т) nH O
6. При некотором скачке потенциала наступает равновесие:
М (т) nH O  М (ж)) ·nH O n .
М (ж)) ·nH O n .
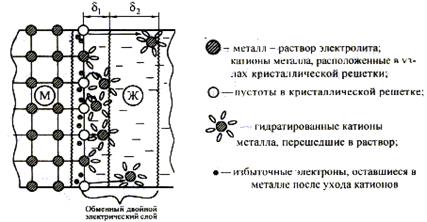
Рис. 12. Схема возникновения обменного двойного электрического слоя и электродного потенциала на границе металл-раствор электролита.
Равновесный электродный потенциал (или просто электродный потенциал) — это скачок потенциала, установившийся между металлом раствором электролита в условиях равновесия.
Величина электродного потенциала, а также толщина двойного электрического слоя зависят от природы металла, температуры и активности (концентрации) ионов в растворе. ДЭС имеет диффузное строение, поэтому абсолютное значение электродного потенциала определить невозможно. Однако, если использовать электрод сравнения, потенциал которого условно принят равным нулю (Е1= 0(В)), можно вычислять электродные потенциалы неизвестных полуэлементов (Е2), относительно электрода сравнения при помощи уравнения: Э.Д.С. (DЕ) = Е1 – Е2.
На практике пользуются относительным (условным ) значением равновесного электродного потенциала , обозначаемым 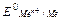 .
.
Стандартный относительный электродный потенциал 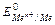 — это величина равная электродвижущей силе электрохимической ячейки, составленной из электрода, опущенного в раствор с активностью ионов металла, равной 1моль/л при 25 0С (298К), при Р=1.03.105 Па, и электрода сравнения.
— это величина равная электродвижущей силе электрохимической ячейки, составленной из электрода, опущенного в раствор с активностью ионов металла, равной 1моль/л при 25 0С (298К), при Р=1.03.105 Па, и электрода сравнения.
В качестве электрода сравнения используют стандартный водородный электрод (рис.2). Конструктивно он состоит из платиновой пластинки (покрытой слоем электролитической платины — платиновой чернью), частично погруженной в раствор, содержащий ионы водорода (обычно раствор H2SO4 или HCl) . К пластинке подается чистый газообразный водород под постоянным давлением. В кислой среде водородному электроду H3O /H2 , Pt отвечает уравнение электродного процесса 2H3O (ж) 2 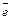 H2 (Г) 2H2O (ж), которое в упрощенном виде записывается: 2Н (ж) 2 H2 (Г).
H2 (Г) 2H2O (ж), которое в упрощенном виде записывается: 2Н (ж) 2 H2 (Г).
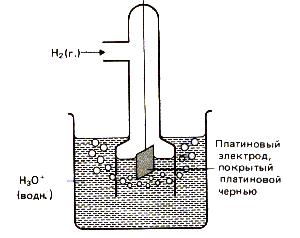
Рис.13. Водородный электрод.
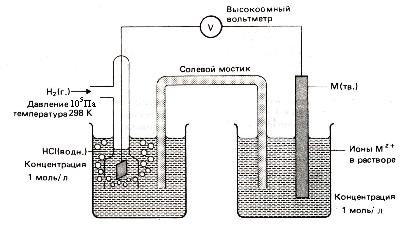
Рис.14. Измерение стандартных электродных потенциалов
В схематической записи водородный электрод представляют так: 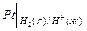 , если платина служит анодом или
, если платина служит анодом или
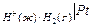 , если платина служит катодом.
, если платина служит катодом.
Использование водородного электрода позволяет измерить относительный электродный потенциал любого полуэлемента, составляя с его помощью химический источник тока (рис.3).
Если водородный электрод играет роль отрицательного электрода, схематическая запись составленного таким образом химического источника тока имеет вид: А— PtêH2(г) êН (ж) êêМn (ж) êМ (т) К . Следовательно,
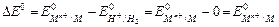 .
.
Если водородный электрод играет роль положительного электрода. А — М (т) êМn (ж) êê Н (ж) ê H2(г) ê Pt К , то 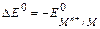 .
.
Таким образом, стандартный электродный потенциал рассматриваемого полуэлемента равен стандартной э.д.с. химического источника, состоящего из этого полуэлемента и стандартного водородного электрода.
В нестандартных условиях электродный потенциал расчитывают по уравнению Нернста:
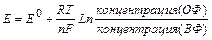
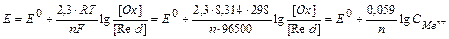
[OX]-концентрация окислителя;
[Red]-концентрация восстановителя;
(ОФ) — окисленная форма;
(ВФ) — восстановленная форма;
R=8.314 Дж/К.моль (универсальная газовая постоянная);
Е0-относительный стандартный электродный потенциал (табличные данные); измеряется в вольтах (В);
Т-абсолютная температура (298К);
F-число Фарадея, равное 96500 (Кл/моль);
n-число электронов, участвующих в процессе;
С-молярная концентрация катионов металла (моль/л).
Е=Е0 0.059/n.lgCMen , если CMen =1 моль/л, то Е = Е0.
Задача. Вычислить электродный потенциал золотой пластины, опущенной в раствор соли с концентрацией [Au3 ], равной 0.1 моль/л.
Решение:
Поскольку условия, описываемые в задаче, отличаются от стандартных, т.е. СAu3 ¹1 моль/л, а составляет 0,1 моль/л по условию задачи, следовательно, для рассчета электродного потенциала необходимо использовать уравнение Нернста:
ЕAu3 /Au=1.5 (0.059/3).lg10-1=1.48 (В).
Понятие стандартный электродный потенциал введено для того, чтобы можно было сравнить окислительно-восстановительные свойства различных систем по их стандартным потенциалам.
В соответствии со значениями стандартного электродного потенциала был составлен электрохимический ряд напряжений металлов.
K Ca Mg Al Zn Fe Pb H Cu Ag Au
-3.0 -2.9 -2.4 -1.7 -0.76 -0.44 -0.13 0 0.34 0.8 1.5
1.Все металлы, стоящие левее водорода , имеют отрицательные значения стандартных электродных потенциалов , правее –положительные .Соглашение о знаках потенциалов было принято в 1953г. на конгрессе Международного союза теоретический и прикладной химии (ИЮПАК) . Условились приписывать потенциалу тот знак , который имеет электрод в паре со стандартным водородным электродом .
2. Отрицательные значения свидетельствуют, что данные металлы являются восстановителями по отношению к водороду, т.е. вытесняют (восстанавливают его из растворов минеральных кислот с 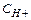 =1 моль/л, анионы которых не проявляют окислительных свойств (HCl , разб.H
=1 моль/л, анионы которых не проявляют окислительных свойств (HCl , разб.H  SO
SO  ))
))
Zn 2HCl→ZnCl H ↑; E0Znn /Zn= -0.763 B
и разлагают воду
2Na 2H O→2NaOH H ↑; E0Na /Na = -2.711 B
3. Чем более отрицательно значение электродного потенциала, тем выше способность металла отдавать свои ионы в раствор, т.е. тем сильнее выражена восстановительная способность его восстановленной формы (способность отдавать электроны) и ниже окислительная способность его окисленной формы (способность принимать электроны).
4.Положительное значение потенциала означает, что данные ионы металлов являются окислителями по отношению к водороду, т.е. не разлагают воду и не вытесняют его из растворов, в которых =1моль/л. Наоборот, водород вытесняет металлы из растворов их солей.
5. Среди металлов с положительным значением стандартного электродного потенциала, каждый предыдущий металл вытесняет все последующие из растворов их солей. Например: Cu HgCl2→CuCl2 Hg.
§
План
1. Работа гальванического элемента (ГЭ).
2. Применение ГЭ. Перспективы развития.
2.1 кислотные аккумуляторы;
2.2 щелочные аккумуляторы;
2.3 топливные элементы.
Литература
1. Фролов В.В. Химия. Гл.V, §51-56.
2. Ахметов Н.С. Общая и неорганическая химия. Раздел V, гл.3,4.
3. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
Химические источники тока (ХИТ) — это гальванические элементы, аккумуляторы и топливные элементы (ТЭ). Их используют как автономные малогабаритные источники энергии для транспортных двигателей и машин, радиотехнических устройств, приборов управления, освещения и т.д. К достоинствам современных ХИТ можно отнести относительно высокие КПД и удельную мощность. Это позволяет им конкурировать с другими видами источников энергии.
Чтобы создавать и правильно использовать ХИТ, необходимо знать закономерности протекания электрохимических процессов, в основе которых лежат реакции ОВР.
Гальваническими элементами (ГЭ)называются устройства, преобразующие энергию ОВР в электрическую. Они обычно состоят из двух электродов (анода и катода), изготовленных из различных металлов или их соединений, погруженных в электролиты (растворы солей этих металлов, растворы кислот, щелочей, токопроводящие пасты).
Анод (А)– это электрод, на котором происходит процесс окисленияили отдачи электронов.
Катод (К)— это электрод, на котором происходит процесс восстановленияили принятия электронов.
Элемент Даниэля-Якоби является одним из самых простых и распространенных гальванических элементов. Он состоит из цинкового и медного электродов, погруженных соответственно в растворы сульфата цинка и сульфата меди. Растворы солей разделены пористой полупроницаемой перегородкой или соединены электролитическим мостиком
1 стадия работы ГЭ:растворение (окисление) электрода, изготовленного из более активного металла (анода)
Zn0 nH2O — 2ē → Zn2 ·nH2O
Zn0 — 2ē → Zn2
2 стадия – это прохождение электрического тока (направленный поток ē) по металлическому проводнику. Если цинк и медь соединить проводником, то электроны будут перемещаться от цинкового анода ( А—) к медному катоду ( К ).
3 стадия-на поверхности катода произойдет разрядка (восстановление) ионов Сu2 :
Cu2 ·nH2O 2ē → Cu0 nH2O
Cu2 2ē → Cu0.
Чтобы ГЭ работал, необходимо, чтобы две изолированные системы были соединены электролитическим мостиком (агар-агар KCl) или полупроницаемой перегородкой, по которым от К к А— будут перемещаться ионы SO42-. Все стадии процессов,протекающих в ГЭ, связаны между собой и идут с одинаковой скоростью.
Схема работы гальванического элемента.
(-) ( )
(А) Zn │ ZnSO4 ││CuSO4 │Cu (K)
если[Zn2 ]=[Cu2 ]=1моль/л
Анодный процесс: Zn0 — 2ē → Zn2 E0A= -0,76 (B)
Kатодный процесс: Cu2 2ē → Cu0 E0K= 0,34 (B)

Ионное уравнение: Zn Cu2 → Zn2 Cu0
SO42- SO42-

Молекулярное уравнение: Zn CuSO4 → ZnSO4 Cu ЭДС=Е0-ЕВ=ЕК-ЕА=0.34-(-0.76)=1.18 (В).
В гальванической паре анодом (А—) всегда будет электрод с меньшим значением электродного потенциала (Е), а катодом (К ) – электрод с большим значением Е.
Работа по переносу электронов во внешней цепи и ионов во внутренней цепи в ГЭ равна произведению перенесенного заряда (n.F) на разность потенциалов (ΔE). Максимальное значение работы (Амах) достигается, когда ГЭ работает обратимо. В этом случае Амах=n.F. ΔE. Для станадартных условий Амах для элемента Даниеля-Якоби составляет:
Амах=n.F. ΔE=2.96500.1,1=212,46 кДж
Максимально полезная работа, которую может совершить система при протекании реакции в условиях постоянного давления и температуры, равна убыли энергии Гиббса (более подробно этот вопрс рассматривается в лекции по термодинамике).
Амах=ΔG0298
ΔG = -nFΔE,
где n – число электронов, участвующих в процессе;
F – число Фарадея;
ΔЕ – ЭДС.
Это уравнение устанавливает связь между химической и электрической энергиями. ЭДС ГЭ подобно ΔG может служить критерием равновесия и возможности самопроизвольного протекания окислительно-восстановительных реакций в определенном направлении: при этом условии – самопроизвольное протекание процесса (ΔG<0) соответствует условию ΔЕ>0. Критерием равновесия (ΔG=0) является равенство ЭДС ГЭ нулю.
С учетом последнего уравнение Нернста приобретает вид:
ЭДС= ΔЕ= (RT/n.F).lnKР,
Где КР— термодинамическая константа равновесия токообразующей реакции. Это выражение позволяет рассчитывать константу равновесия КР любой токообразующей ОВР.
Задача. Будет ли работать ГЭ, состоящий из никелевого электрода, погруженного в раствор NiSO4 с концентрацией 0,01 моль/л и стандартного железного электрода? Составьте схему, напишите уравнения электродных процессов, рассчитайте ЭДС и ΔG0298 элемента и сделайте вывод о возможности его работы.
Решение. Определяем потенциал электродов. Для стандартного железного электрода потенциал находим в ряду напряжений металлов. 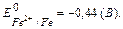 Для никелевого электрода вычисляем потенциал по уравнению Нернста.
Для никелевого электрода вычисляем потенциал по уравнению Нернста.
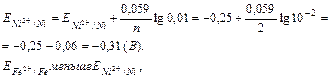
следовательно в ГЭ анодом является железо, а катодом — никель.
Запишем схему ГЭ:
(-) ( )
(А) Fe │ FeSO4 ││ NiSO4 │ Ni (K)
-0,44(В) 1моль/л 10-2 моль/л -0,31(В)
Запишем уравнения электродных процессов:
А: Fe0 — 2ē → Fe2 окисление ЕА=-0,44 (В)
K: Ni2 2ē → Ni0 восстановление ЕК=-0,31 (В)

Fe Ni2 → Fe2 Ni0
SO42- SO42-
Fe NiSO4 → FeSO4 Ni
ЭДС=Е0-ЕВ=-0,31 0,44=0,13(В)
Для вычисления изменения энергии Гиббса воспользуемся формулой:
ΔG = -nFΔE= -2∙96500∙0,13 = -25090 Дж = -25,09 КДж, ΔG<0.
Этот ГЭ будет работать, хотя и имеет малую ЭДС.
Аккумуляторы– это химические источники тока многократного действия, их можно регенерировать, пропуская через них постоянный электрический ток.
Свинцовые аккумуляторы (кислотные) изготавливают из свинцовых пластин, на которые нанесен оксид свинца (PbO).
А Pb│ H2SO4│PbO К
Когда аккумулятор заливают серной кислотой, идет реакция:
PbO H2SO4 → PbSO4 H2O
Далее аккумулятор заряжают от внешнего источника постоянного тока.
 Зарядка (электролиз)
Зарядка (электролиз)
К — Pb2 SO4 2ē → Pb0 SO42-(на К— восстановление)
A Pb2 SO4 — 2ē 2H2O → Pb 4O2 SO42- 4H (на А окисление)

2PbSO4 2H2O → Pb PbO2 2H2SO4
Заряженный аккумулятор можно сразу использовать по назначению. При хранении из аккумулятора надо вылить электролит, промыть водой и хранить до двух лет. Для дальнейшего использования надо залить электролит.
Разрядка.При разрядке анод и катод меняются местами.
Идут следующие реакции:
А—Pb0 SO42- — 2ē → PbSO4 1
К PbO2 2ē SO42- 4H → PbSO4 2Н2О 1

Pb 2H2SO4 PbO2 → 2PbSO4 2H2O
ЭДС=2,1 В.
Для промывки аккумулятора готовят раствор, в котором на 1 л 5% NH4OH берут 75 мл 2% трилона Б.
Преимущества свинцового аккумулятора:
1) большая электрическая емкость;
2) устойчивость в работе;
3) большое количество циклов зарядка-разрядка.
Недостатки:
1) большая масса;
2) выделение водорода при зарядке;
3) не герметичность при наличии концентрированного раствора серной кислоты.
Кадмиево-никелевые аккумуляторы (щелочные)
В основе их работы лежит химическая реакция:
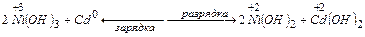 ЭДС = 1,4 В
ЭДС = 1,4 В
В аккумулятор заливают щелочной электролит, который готовят следующим образом : на 0,5 л воды берут 130 г КОН и 10 г LiOH . При зарядке идут следующие процессы:
К — Cd2 (OH)2 2ē → Cd0 2OH— 1
A 2Ni2 (OH)2 2OH — 2ē → 2Ni 3OOH— 2H2O 1
Cd2 (OH)2 2Ni 2(OH)2 Cd0 2Ni 3OOH 2H2O
Cd0 2Ni 3OOH 2H2O
При разрядке:
А— Cd0 — 2ē 2OH— → Cd(OH)2
K Ni2 3O3∙H2O → 2Ni 3OOH 2H2O 2ē → 2Ni(OH)2 2OH—

разрядка
 Cd 2NiOOH 2H2O Cd(OH)2 2Ni(OH)2
Cd 2NiOOH 2H2O Cd(OH)2 2Ni(OH)2
 зарядка
зарядка
В принципе, электрическую энергию может дать любая ОВР. Однако, число реакций, практически используемых в химических источниках электрического тока, невелико. Это связано с тем, что не всякая ОВР позволяет создать гальванический элемент, обладающий технически ценными свойствами (высокая и постоянная ЭДС, длительная сохранность и др.), кроме того, многие ОВР требуют расхода дорогостоящих веществ. В современных ГЭ (это устройства однократного действия) и аккумуляторах (устройства многократного действия) используют не 2, а 1 электролит. В качестве анода чаще всего используют цинк, а в качестве катода оксиды менее активных металлов. Все химические источники тока имеют два существенных недостатка:
1) высокую стоимость веществ (свинец, кадмий), из которых их изготавливают;
2) малое отношение количества энергии, которую дает элемент к его массе.
Работая над этой проблемой, пришли к созданию топливных элементов.
§
В них отрицательный электрод состоит из порошкообразного железа, спресованного с небольшим количеством специальных добавок (оксида ртути и др.), а положительный электрод – из гидроксида никеля (III), в который для повышения электропроводности добавляют чистый графит. Электролитом служит 23% раствор едкого кали (ρ = 1,21г/см3).
Разрядка:
А—Fe — 2ē → Fе 1
K Ni(OH)3 ē → Ni(OH)2 OH— 2

Fe 2Ni(OH)3 ↔ 2Ni(OH)2 Fe(OH)2
ЭДС = 1,35 (В).
При зарядке идет обратный процесс.
Топливные элементы
В них в качестве топлива используют уголь, кокс, природные и искусственные горючие газы , а в качестве окислителя – О2, воздух.Примером может служить щелочной водородно-кислородный элемент. Его электроды изготавливают из полых, пористых трубок, на которые наносят катализаторы. На водородный электрод платину; на кислородный электрод сплавы Co Al или Fe Mn Ag.
По трубкам пропускают ток водорода и кислорода. Работу такого элемента можно описать следующими уравнениями:
А— 2H2 4OH — — 4ē → 4H2O EA=-0,83 B
К O2 2H2O 4ē → 4OH— EK= 0,4 B

O2 2H2O 2H2 4OH — → 4OH — 4H2O
O2 2 H2O → 2H2O
ΔG = -574 КДж.
ЭДС=ЕК-ЕА=0,4 0,83=1,23 В
Такие топливные элементы используются в спутниках, космических кораблях, военной технике.
§
План
1. Определение понятия «коррозия металлов».
2. Классификация коррозионных процессов
2.1. по виду изменения поверхности металла;
2.2. по механизму;
2.3. по характеру дополнительных воздействий.
3. Химическая коррозия, ее виды. Химизм.
4. Электрохимическая коррозия, ее виды и химизм.
5. Факторы, влияющие на скорость коррозии.
6. Защита металлов от коррозии.
Литература:
1. Фролов В.В. Химия. Гл.V, §51-56.
2. Лучинский Г.П. Курс химии. Гл.V, §8-12, гл. VI, §13-18
3. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
4. Абраменко В.Л. Методические указания к самостоятельному изучению темы “Коррозия и защита металлов от нее”. Луганск, 1991 г.
Металлы вследствие своей высокой прочности, пластичности, износоустойчивости, тепло- и электропроводности являются наиболее важными конструкционными материалами.
В процессе эксплуатации в результате воздействия окружающей среды происходит их разрушение, так называемая коррозия.
Потери от коррозии в ведущих индустриальных странах составляют около 3-5% валового национального продукта, а затраты на возмещениекоррозионных потерь во всем мире исчисляются сотнями миллиардов долларов, поэтому раздел «Коррозия металлов и методы защиты их от коррозии» является одним из важнейших в курсе химии для инженерных специальностей.
Обычно корродируют металлы, которые встречаются в природе не в самородном состоянии, как Au, Pt, а в виде различных руд. На извлечение этих металлов из природных соединений расходуется значительное количество энергии (Ме n nē → Me0; ΔG>0), которая накапливается в металлах, делая их термодинамически неустойчивыми, химически активными веществами (Ме0 — nē → Me n; ΔG<0). В результате такого самопроизвольно протекающего коррозионного процесса металлы переходят в оксиды, гидроксиды, карбонаты, сульфиды и другие соединения и теряются безвозвратно.
Итак, коррозию можно определить как самопроизвольно протекающий окислительно-восстановительный процесс (ΔG<0) разрушения металла под воздействием окружающей среды, происходящий с выделением энергии (ΔН<0) и рассеиванием вещества (ΔS>0).
Механическое разрушение металлов, происходящее по физическим причинам, не называют коррозией, а называют эрозией, истиранием, износом.
По характеру разрушения поверхности коррозию подразделяют на сплошную и местную. Сплошная коррозия подразделяется на равномерную, если процесс окисления происходит по всей поверхности металла с одинаковой скоростью, и неравномерную – процесс окисления происходит по всей поверхности с различной скоростью на различных участках металла.
Местная коррозия подразделяется на коррозию пятнами, точечную, питтинг (углубленно-точечную), межкристаллитную(наиболее опасна, т.к. ослабляет связи между зернами структуры сплавов), растрескивающуюся, селективную (избирательную).
По механизму протекания различают следующие виды коррозии:
— химическая (газовая, жидкостная);
— электрохимическая (концентрационная, контактная, электрокоррозия);
— особые виды (биологическая, радиационная, ультразвуковая).
По характеру дополнительных воздействий различают:
— коррозию под влиянием механических напряжений;
— коррозию при трении;
— кавитационную коррозию (возникает при одновременном коррозионном и ударном воздействии агрессивной среды, когда лопаются пузырьки воздуха при работе лопастей гребного винта, роторов насосов).
Рассмотрим более подробно виды коррозии по механизму протекания.
Под химической коррозией понимают разрушение металлов окислением в окружающей среде без возникновения электрического тока в системе.
Газовая коррозия протекает при обычных условиях, но чаще при высоких температурах. Наблюдается при разливе расплавленных металлов, их термической обработке, ковке, прокатке, сварке и т.д.
Самый распространенный случай газовой коррозии – взаимодействие металла с кислородом:
xMe 1/2yO2 → MexOy
4Fe 3O2 → 2Fe2O3
Образующаяся при такой коррозии оксидная пленка в ряде случаев играет защитную функцию. Для этого она должна быть сплошной, беспористой, иметь хорошее сцепление с металлом, обладать твердостью, износостойкостью и иметь коэффициент термического расширения, близкий к этой величине для металла. Все эти качества оксидной пленки можно оценить по фактору Пиллинга-Бэдвордса (a). Металлы (щелочные, щелочноземельные), у которых a<1 имеют тонкие оксидные пленки с разрывами. Сплошные и устойчивые оксидные слои образуются при a=1,2 – 1,6 (например, Al2O3, ZnO, NiO и т.д.). При значениях a значительно больше единицы пленки получаются неслошные, лекго отделяющиеся от поверхности металла (железная окалина).Коррозионно-активными газами, кроме кислорода, являются: угарный газ, углекислый газ, сернистый ангидрид, азот, его оксиды и галогены. Например, при разливе расплавленного алюминия, происходит его взаимодействие не только с кислородом, но и с азотом воздуха.
4Al 3O2 → 2Al2O3; 2Al N2 → 2AlN.
Жидкостная коррозия протекает, как правило, в жидких неэлектролитах: спиртах, хлороформе, бензоле, бензине, керосине и других нефтепродуктах. Ускоряет процесс жидкостной коррозии сера,кислород, галогены, влага, атакже повышенная температура (коррозия поршней в двигателях внутреннего сгорания),что можно описать уравнениями : Me(II) R1 – S – R2 → MeS R1 – R2
Me(I) nR – Cl → MeCl 1/2nR – R ,
где R1 – S – R2и nR – Cl углеводороды, содержащие серу и хлор.
Электрохимическая коррозия наиболее распространенный вид коррозии. Это разрушение деталей, машин, конструкций в грунтовых, речных, морских водах, под влиянием воды (росы), под воздействием смазочно-охлаждающих жидкостей, используемых при механической обработке металлов, атмосферная коррозия и т.д.
Электрохимическая коррозия – это пространственно разделенный окислительно-восстановительный процесс разрушения металла, протекающий в среде электролита, с возникновением внутри системы электрического тока, называемого коррозионным током.
Рассмотрим химизм атмосферной коррозии стального изделия. Сталь – это сплав железа с углеродом, в котором углерода менее 2%, например, цементит (Fe3C4). При электрохимической коррозии во влажном воздухе (О2 2Н2О) железо и цементит образуют микрогальванопару, в которой роль анода выполняет железо, а цементит – роль катода.
Схема процесса:
(А) Fe │H2O, O2 │ Fe3C4 (K)
Анодный процесс: Fe0 — 2ē → Fe2 2 поляризация
Катодный процесс: 2H2O O2 4ē → 4OH— 1 деполяризация

Суммарное уравнение коррозионного процесса разрушения стального изделия, находящегося во влажном воздухе:
2Fe 2H2O O2 → 2Fe(OH)2
Для железа более характерна степень окисления (3 ), поэтому процесс окисления идет дальше:
4Fe(OH)2 2H2O O2→4Fe(OH)3,
образующийся Fe(OH)3 при нагревании может терять воду.
Fe (OH)3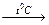 H2O FeOOH.
H2O FeOOH.
То есть продуктами коррозии железа (ржавчина) является смесь различных соединений. Если учесть, что в воздухе присутствуют углекислый газ, сернистый газ, следовательно, могут образовываться и соли железа.
 Часто из-за различной рельефности металлических конструкций, в том числе и стальных, на некоторых участках скапливается вода, при этом происходит так называемая концентрационная коррозия, обусловленная различной концентрацией деполяризатора кислорода (в случае атмосферной коррозии), водорода (в кислой среде) на различных участках металла. Там, где концентрация деполяризатора больше (края капли воды), формируется катодный участок, где концентрация деполяризатора меньше (центр капли воды) – анодный участок (рис.15).
Часто из-за различной рельефности металлических конструкций, в том числе и стальных, на некоторых участках скапливается вода, при этом происходит так называемая концентрационная коррозия, обусловленная различной концентрацией деполяризатора кислорода (в случае атмосферной коррозии), водорода (в кислой среде) на различных участках металла. Там, где концентрация деполяризатора больше (края капли воды), формируется катодный участок, где концентрация деполяризатора меньше (центр капли воды) – анодный участок (рис.15).
После высыхания капли в её центре обнаруживается углубление, а иногда даже и отверстие (для пластин толщиной 0,1 …0,2 мм). Такие процессы часто наблюдаются при атмосферной и почвенной коррозии железных и стальных изделий (троса, стопки листов и т.д.) – точечная коррозия, переходящая в питтинг. Следует отметить, что хотя конечный продукт коррозии (ржавчина) нерастворим, однако он не препятствует процессу растворения металла, поскольку формируется за пределами анодного участка (на границе соприкосновения его с катодами) в виде кольца внутри капли.
На практике часто встречаются случаи, когда металлы различной активности находятся в контакте друг с другом, образуя гальванопары. Кроме того, технические металлы содержат примеси других металлов, сплавы содержат различные металлы. Такой металл или сплав, находясь в среде электролита, дает множество микро — и макрогальванопар, в которых анодом является более активный металл, т.е. металл с меньшим значением электродного потенциала, именно он и подвергается коррозии.
Рассмотрим случай контактной коррозии с водородной деполяризациейцинка и меди, в сернокислой среде. Цинк и медь, имеют различные значения электродных потенциалов. Более активным в этой гальвано паре является цинк (Е0Zn2 /Zn = -0,76 В), он имеет меньшее значение электродного потенциала и будет анодом, т. е именно цинк будет подвергаться коррозионным процессам, менее активным металлом является медь (Е0Cu2 /Cu = 0,34 В), она будет катодом.
Запишем схему: (А) Zn │ H2SO4 │ Cu (K)
Анодный процесс: Zn0 — 2ē → Zn2
Катодный процесс: 2Н 2ē → Н2 деполяризатор

Суммарное ионное уравнение: Zn 2H → Zn2 H2
Молекулярное уравнение: Zn H2SO4 → ZnSO4 H2.
Факторы, влияющие на скорость коррозии:
а) напряжение и деформация при механической обработке металлов;
б) перемешивание агрессивной среды;
в) дифференциальная аэрация;
г) температура;
д) кислотность среды (рН).
Рассматривая фактор (д) обратите внимание, что электродные потенциалы металлов существенно зависят от состава электролита и рН среды. Так, в случае контактной (Al-Zn) коррозии в 1М растворе HCl
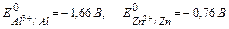 ,
,
возникает гальвано пара, в которой роль анода выполняет Al, а катода- Zn, схема такого процесса: (А) Al │ HCl │ Zn (K)
1моль/л
В 0,1 М растворе HCl 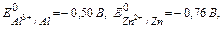 в этом случае большую активность имеет цинк, он будет в гальвано паре анодом, алюминий – катодом, а схему запишем так: (А) Zn │ HCl │ Al (K)
в этом случае большую активность имеет цинк, он будет в гальвано паре анодом, алюминий – катодом, а схему запишем так: (А) Zn │ HCl │ Al (K)
0,1моль/л
Электрокоррозия – протекает под действием блуждающих токов, возникает от постоянных источников тока (электротранспорт, трансформаторы, линии электропередач). Рассматривая коррозию под действием блуждающих токов, надо помнить, что место выхода тока – будет анодным участком, входа тока – катодным, участок протекания тока – нейтральной зоной. Радиус действия блуждающих токов может достигать нескольких десятков километров. Ток силой 1А за год разрушает до 3 кг алюминия, 9 кг железа, 11 кг цинка или меди, 34 кг свинца.
ЗАЩИТА МЕТАЛЛОВ ОТ КОРРОЗИИ
Потери от коррозии в мировой экономике огромны. Около 1/3 вводимого в эксплуатацию металла подвергается коррозии, при этом примерно 10% теряется безвозвратно.
Борьба с коррозией осуществляется различными методами. Наиболее рациональный и надежный путь – изготовление аппаратов и машин изкоррозионно-стойких металлических или неметаллических материалов,но из-за дороговизны таких материалов, чаще используют дешевые и доступные металлы с последующей защитой их от коррозии. Полностью избежать коррозии невозможно, но, применив определенные методы защиты, можно снизить ее воздействие.
Можно условно выделить следующие группы методов защиты металлов от коррозии:
1.Создание рациональных конструкций, т.е. таких, которые не имеют застойных зон и других мест скопления влаги, грязи и других коррозионно-агрессивных сред, допускают быструю очистку и аэрацию.
2.Легирование металлов. Это эффективный, хотя обычно дорогой метод повышения коррозионной стойкости металлов. При легировании в состав сплава вводят компоненты (Cr, Ni, W, Si, V, Mo, Re и другие), вызывающие пассивирование металла. Механизм защиты (например, в нержавеющих сталях) состоит в образовании на поверхности плотных оксидных слоев, типа шпинелей состава NiO . Cr2O, FeO . Cr2O3, которые оказываются более устойчивыми, чем просто оксиды хрома или никеля.
3.Создание аморфных структур металлов. Путь к этому способу защиты открыла сверхбыстрая закалка. Расплавленный металл из тигля подают в тончайший зазор между двумя массивными валками и подвергают формированию и резкому охлаждению. В этих условиях атомы не успевают выстраиваться в присущие металлам кристаллические решетки, фиксируется «хаос атомов», свойственный расплавленному металлу. В результате получается аморфная структура, подобная стеклу, резко возрастает коррозионная устойчивость металлов.
4.Защитные покрытия – самый распространенный метод защиты металлов от коррозии. Смысл их нанесения – изоляция от агрессивной среды. Различают неметаллические и металлические покрытия.
а) неметаллические покрытия получают нанесением на поверхность металла лака, краски, смолы, олифы, эмали или стеклоэмали. Поверхность металла покрывают также резиной, эбонитом, полимерными материалами, цементом, бетоном, оксидными пленками: ZnO, Al2O3 (оксидирование) и нитридными пленками: Fe4N, Fe2N (азотирование). Покрыть поверхность металла можно осаждением нерастворимых фосфатов этого металла: Fe(H2PO4)2 2 Fe2 ® Fe3(PO4)2¯ 4H (фосфатирование) или насыщением поверхности металла углеродом (цементация).
б) защитные покрытия металлами. Для этого используют коррозионно-устойчивые металлы (Sn, Zn, Al, Au, Ag, Ni, Cr и др.) Различают анодные и катодные металлические покрытия. Если защищаемый металл покрывают более активным металлом, то такое покрытие называют анодным. При нарушении покрытия разрушается металл покрытия. Рассмотрим это на примере оцинкованного железа. Составим схему коррозионного разрушения.
( — ) ( )
(А) Zn │ H2O, O2 │ Fe (K)
A: Zn0 — 2ē → Zn2 2
K: 2H2O O2 4ē → 4OH— 1

2Zn 2H2O O2 → 2Zn(OH)2
Если защищаемый металл покрыт менее активным металлом, например, железо покрыто оловом, то такой вид покрытия называется катодным. При нарушении покрытия разрушается основной металл. Рассмотрим этот случай коррозии.
( — ) ( )
(А) Fe │ 2H │ Sn (K)
A: Fe0 — 2ē → Fe2 1
K: 2H 2ē → H2↑ 1
Fe 2H → Fe2 H2
5. Электрохимические методы защиты:
а) защита внешним потенциалом);
б) анодная (протекторная).
Защита внешним потенциалом (чаще катодная) осуществляется подключением защищаемой конструкции к отрицательному полюсу (катоду) внешнего источника тока с очень малым напряжением (0,1 В). К положительному полюсу подсоединяется лом, который и разрушается. Этот вид защиты используют для металлических сооружений: трубопроводов, резервуаров и т.д.
Протекторная защитазаключается в том, что к изделию, подвергающемуся электрохимической коррозии, подключают деталь – протектор из более активного металла, чем металл изделия. Протектор будет разрушаться, а изделие останется неизменным. Применяют в паровых котлах, для защиты корпусов морских и речных судов, трубопроводов, рельсов и т.д.
Задача. Приведите пример протекторной защиты в электролите, содержащем растворенный кислород. Составьте уравнения анодного и катодного процессов и вычислите ЭДС реакции.
 Решение. Протекторная защита осуществляется путем присоединения к железу более активного металла, обычно цинка, магния и их сплавов. Таким образом, создается искусственный микрогальванический элемент. Чаще всего используют протекторную защиту в растворах электролитов (паровые котлы, химические аппараты), в морской воде и в почве (защита трубопроводов). Рассмотрим протекторную защиту от почвенной коррозии:
Решение. Протекторная защита осуществляется путем присоединения к железу более активного металла, обычно цинка, магния и их сплавов. Таким образом, создается искусственный микрогальванический элемент. Чаще всего используют протекторную защиту в растворах электролитов (паровые котлы, химические аппараты), в морской воде и в почве (защита трубопроводов). Рассмотрим протекторную защиту от почвенной коррозии:
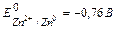
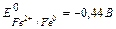
Среда нейтральная или слабощелочная, так как концентрация солей невелика. В этом, созданном нами, коррозионном элементе анодом служит протектор (цинк), он растворяется.
Анод: Zn0 – 2 ® Zn2 .
Электроны передаются на железо. Деполяризатором в этом случае является кислород.
Катод: O2 2H2O 4 ® 4OH—; E0 = 0,40 B.
Суммарная реакция: 2Zn0 O2 2H2O = 2Zn(OH)2.
ЭДС реакции определяем: DЕ = Екатода – Еанода = 0,40 – (-0,75) = 1,16 В.
Ответ: протектор Zn, он окисляется и защищает железо; DЕ=1,16 В.
6. Воздействие на агрессивную среду. Для замедления коррозии в агрессивную среду вводят вещества, называемые ингибиторами (замедлителями). Это чаще всего органические вещества, пассивирующие поверхность металла: тиомочевина C(NH2)2S, диэтиламин C2H5— NH — C2H5, уротропин (CH2)6N4, неорганические вещества SiO32-, NO2—, Cr2O72-, а также освобождение воды от растворенного в ней кислорода (воду фильтруют через слой железных опилок). Либо удаляют активаторы коррозии, например, ионы Cl—, Br—, F—, SO42-, NO3—.
ЭЛЕКТРОЛИЗ.
План
1. Сущность процессов протекающих при электролизе.
2. Виды электролиза:
2.1 электролиз расплавов,
2.2 электролиз водных растворов.
3. Последовательность разрядки ионов на электродах.
4. Явление перенапряжения.
5. Законы электролиза.
6. Применение электролиза.
Литература.
1. Фролов В.В. Химия. Гл.V, §51-56.
2. Лучинский Г.П. Курс химии. Гл.V, §8-12, гл. VI, §13-18
3. Общая химия под ред. Соколовской Е.М. и др. Гл.6, §1-11.
§
§
Возможные анодные процессы при электролизе водных растворов:
— окисление молекул воды (рН < 7): 2Н2О — 4ē → О2 4Н ; Е0= 1,23 (В)
— окисление ОН  ионов (рН ≥ 7) : 4ОН— — 4ē → О2 2Н2О;Е0= 0,40 (В)
ионов (рН ≥ 7) : 4ОН— — 4ē → О2 2Н2О;Е0= 0,40 (В)
— анионы кислот по отношению к электроокислению делят на 2 группы:
а) анионы кислород не содержащих кислот (Cl—, Br—, I—, S2—) легко окисляются: 2I— I2; E0 = 0,54 (B);
I2; E0 = 0,54 (B);
б) анионы кислород содержащих кислот (SO42-, NO3—, CO32-, PO43-) удерживают электроны прочнее, чем молекулы воды, поэтому на аноде окисляется вода по приведенной выше схеме.
— растворение анода:. M0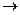 М n n ē
М n n ē
Различают растворимые электроды (Cu;Zn и т.д.) и инертныеэлектроды (C; Au; Pt).
Рассмотрим пример электролиза раствора хлорида меди (ll) с растворимым медным анодом:
CuCl2 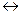 Cu2 2Cl—
Cu2 2Cl—
К — Cu2 (E0 = 0,34 В) А (Cu) Cl—( E0 = 1,36 B)
H2O (E0 = — 0,83 В) H2O (E0 = 1,23)
Cu0 (E0 = 0,34 )
восстанавливаетсяся частица окисляется частица с
с большим потенциалом меньшим потенциалом.
Cu2 Cu0 Cu0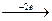 Cu2
Cu2
Этот способ используют для электролитического рафинирования металлов (очистка) в промышленных условиях.
Для того, чтобы произошла разрядка ионов на электродах, к ним надо приложить определенную электродвижущую силу, равную разности потенциалов, называемую потенциалом разложения (Е0разл.), который численно равен по модулю электродвижущей силе гальванического элемента, составленного на основе продуктов электролиза. Например:
рассчитать потенциал разложения 1М соляной кислоты.
HCl ↔ H Cl—
окислитель К— 2Н 2ē → Н2 восстановление
восстановительА 2Cl— — 2ē → Cl2 окисление.
ЭДС = ЕК0 – ЕА0 = 0 – 1,36 =│1,36│В.
Последовательность разрядки ионов при электролизе может нарушаться из-за явления называемого перенапряжением. Перенапряжение равно разности между напряжением положенным на электроды и ЭДС гальванического элемента, отвечающего обратной реакции. Поэтому, делая расчеты реальных электрохимических процессов, необходимо помнить, что реальная разность потенциалов при электролизе (ΔЕ) слагается из: потенциала разложения (Еразл.), ЭДС для преодоления сопротивления электролита, суммы катодного и анодного перенапряжения, которые обусловлены побочными процессами, происходящими на электродах, такими как:
— подход иона к электроду;
— потеря ионом гидратной оболочки (в растворе);
— разрядка иона;
— рекомбинация атомов в молекулы;
— отход молекулы от электрода.
ΔЕразл. = Еразл. IR ΔЕполяриз. ΔЕконтакт.,,
где I – сила тока при электролизе;
R – сопротивление систем;
ΔЕполяриз. – разность потенциалов поляризации, препятствующая прохождению тока;
ΔЕконт. – сумма контактных потенциалов.
R – общее сопротивление системы, равное сумме сопротивлений электролита, диафрагмы электролизера и токоподводов к электролизеру.
Опыт показывает, что потенциалы выделения Е0(Men ) примерно равны их электродным потенциалам, а Е0выделения газов значительно больше их электродных потенциалов. Это объясняется так называемой газовой поляризацией, т.е. образованием на электродах «газовой шубы» за счет выделения водорода на катоде, а кислорода на аноде, или других газов. Газовая поляризация может остановить процессы, протекающие при электролизе, из-за изоляции электродов. Её негативное влияние может быть устранено добавлением окислителей водорода:K2Cr2O7, KMnO4 и восстановителей кислорода Na2SO3, Na3PO4 .
Кроме газовой поляризации, различают:
— концентрационную – за счет изменения концентрации в
приповерхностном слое электрода, если анод медный, то [Cu2 ] у анода больше, чем у катода. Для устранения такой поляризации, необходимо перемешивать электролит;
— химическую– возникает вследствие того, что выделение продуктов
электролиза приводит к образованию гальванической цепи (идет обратная электролизу реакция).
Количественные соотношения при электролизе установлены в 1827г английским физиком Майклом Фарадеем.
Законы Фарадея:
§
m =k . Q; Q=I·t ;
Q-количество электричества, А . с;
I-сила тока, А;
t-время ,с ,ч;
k — электрохимический эквивалент , г/Кл.
Масса вещества, выделившегося при прохождении через электролит одного кулона электричества, называется электрохимическим эквивалентом
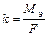 ;
; 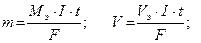 где
где
m – масса вещества, г;
V – объем вещества, л;
Мэ – молярная масса эквивалента, г/моль;
Vэ — эквивалентный объем, л/моль;
F – число Фарадея 96500 Кл/моль, 26,8 А·ч/моль.
II закон: при прохождении через различные электролиты одинакового количества электричества на электродах выделяются массы веществ, пропорционально молярным массам их эквивалентов(химические эквиваленты пропорциональны электрохимическим).
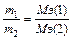
Вследствие протекания побочных процессов на электродах, образуется реально меньшее количество вещества, чем теоретически рассчитанное по вышеуказанной формуле. Для характеристики истинного количества вещества, выделяющегося на электродах, введено понятие выход по току (h).
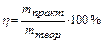 .
.
Задача. Составить схему электролиза раствора сульфата натрия. Написать уравнения катодного и анодного процессов, рассчитать количество образовавшихся при электролизе веществ (для газообразных – объем), если выход по току 70%, а через раствор пропускали ток силой 2А в течение 10 минут.
Сульфат натрия в растворе диссоциирует
Na2SO4 ↔ 2Na SO42-
H2O
На катоде идет восстановление тех частиц, потенциал которых больше, 
поэтому на катоде будет восстанавливаться вода по схеме:
К—: 2НОН 2ē → Н2↑ 2ОН —
к аноду направляются ионы SO42- и H2O.
На аноде окисляются те частицы, електрдный потенциал которых имеет меньшее значение,
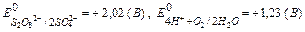 , поэтому будет идти окисление воды по схеме:
, поэтому будет идти окисление воды по схеме:
А : 2Н2О — 4ē → О2 4Н .
Суммарное уравнение электролиза Na2SO4
2Na2SO4 6H2O  2H2 4NaOH O2 2H2SO4
2H2 4NaOH O2 2H2SO4
на катоде у катода на аноде у анода
По закону Фарадея рассчитаем объемы V(Н2), V(О2) и массы m (NaOH), m (H2SO4), зная, что выход по току (ή) составляет 70%.
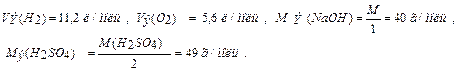
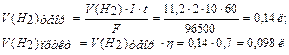
 определяем по второму закону Фарадея:
определяем по второму закону Фарадея:
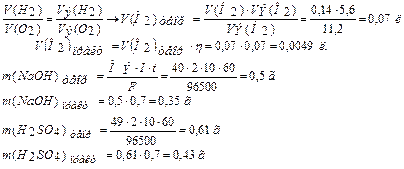
Расчет кислорода в баллонах
Параметры и размеры кислородных баллонов можно посмотреть по ГОСТ 949-73 «Баллоны стальные малого и среднего для газов на Рр ≤ 19,7МПа». Наиболее популярными являются баллоны объемами 5, 10 и 40 литров.
По ГОСТ 5583-78 «Кислород газообразный технический и медицинский» (приложение 2), объем газообразного кислорода в баллоне (V) в кубических метрах при нормальных условиях вычисляют по формуле:
V = K1•Vб,
Vб — вместимость баллона, дм3;
K1 — коэффициент для определения объема кислорода в баллоне при нормальных условиях, вычисляемый по формуле
К1 = (0,968Р 1) * *
Р — давление газа в баллоне, измеренное манометром, кгс/см2;
0,968 — коэффициент для пересчета технических атмосфер (кгс/см2) в физические;
t — температура газа в баллоне, °С;
Z — коэффициент сжигаемости кислорода при температуре t.
Значения коэффициента К1 приведены в таблице 4, ГОСТ 5583-78.
Посчитаем объем кислорода в самом распространенном баллоне в строительстве: объемом 40л с рабочим давлением 14,7МПа (150кгс/см2). Коэффициент К1 определяем по таблице 4, ГОСТ 5583-78 при температуре 15°С:
V = 0,159 • 40 = 6,36м3
Вывод (для рассматриваемого случая): 1 баллон кислорода = 40л = 6,36м3
Таблица 4. ГОСТ 5583-78.
Температура газа в баллоне, °С | Значение коэффициента Ki при избыточном давлении, МПа (кгс/см2) | ||||||||||||||
| 13,7 (140) | 14,2 (145) | 14,7 (150) | 15,2 (155) | 15,7 (160) | 16,2 (165) | 16,7 (170) | 17,2 (175) | 17,7 (180) | 18,1 (185) | 18,6 (190) | 19,1 (195) | 19,6 (200) | 20,1 (205) | 20,6 (210) | |
| -50 | 0,232 | 0,242 | 0,251 | 0,260 | 0,269 | 0,278 | 0,286 | 0,296 | 0,303 | 0,311 | 0,319 | 0,327 | 0,335 | 0,342 | 0,349 |
| -40 | 0,212 | 0,221 | 0,229 | 0,236 | 0,245 | 0,253 | 0,260 | 0,269 | 0,275 | 0,284 | 0,290 | 0,298 | 0,305 | 0,312 | 0,319 |
| -35 | 0,203 | 0,211 | 0,219 | 0,226 | 0,234 | 0,242 | 0,249 | 0,257 | 0,264 | 0,272 | 0,278 | 0,286 | 0,293 | 0,299 | 0,306 |
| -30 | 0,195 | 0,202 | 0,211 | 0,217 | 0,225 | 0,232 | 0,239 | 0,248 | 0,253 | 0,261 | 0,267 | 0,274 | 0,281 | 0,288 | 0,294 |
| -25 | 0,188 | 0,195 | 0,202 | 0,209 | 0,217 | 0,223 | 0,230 | 0,238 | 0,243 | 0,251 | 0,257 | 0,264 | 0,270 | 0,277 | 0,283 |
| -20 | 0,182 | 0,188 | 0,195 | 0,202 | 0,209 | 0,215 | 0,222 | 0,229 | 0,235 | 0,242 | 0,248 | 0,255 | 0,261 | 0,267 | 0,273 |
| -15 | 0,176 | 0,182 | 0,189 | 0,196 | 0,202 | 0,208 | 0,215 | 0,221 | 0,227 | 0,234 | 0,240 | 0,246 | 0,252 | 0,258 | 0,263 |
| -10 | 0,171 | 0,177 | 0,183 | 0,189 | 0,195 | 0,202 | 0,208 | 0,214 | 0,220 | 0,226 | 0,232 | 0,238 | 0,244 | 0,250 | 0,255 |
| -5 | 0,165 | 0,172 | 0,178 | 0,184 | 0,190 | 0,195 | 0,202 | 0,207 | 0,213 | 0,219 | 0,225 | 0,231 | 0,236 | 0,242 | 0,247 |
| 0 | 0,161 | 0,167 | 0,172 | 0,179 | 0,184 | 0,190 | 0,196 | 0,201 | 0,207 | 0,213 | 0,219 | 0,224 | 0,229 | 0,235 | 0,240 |
| 5 | 0,157 | 0,162 | 0,168 | 0,174 | 0,179 | 0,185 | 0,190 | 0,196 | 0,201 | 0,207 | 0,212 | 0,217 | 0,223 | 0,228 | 0,233 |
| 10 | 0,153 | 0,158 | 0,163 | 0,169 | 0,174 | 0,180 | 0,185 | 0,191 | 0,196 | 0,201 | 0,206 | 0,211 | 0,217 | 0,222 | 0,227 |
| 15 | 0,149 | 0,154 | 0,159 | 0,165 | 0,170 | 0,175 | 0,180 | 0,186 | 0,191 | 0,196 | 0,201 | 0,206 | 0,211 | 0,216 | 0,221 |
| 20 | 0,145 | 0,150 | 0,156 | 0,160 | 0,166 | 0,171 | 0,176 | 0,181 | 0,186 | 0,191 | 0,196 | 0,201 | 0,206 | 0,211 | 0,215 |
| 25 | 0.142 | 0,147 | 0,152 | 0,157 | 0,162 | 0,167 | 0,172 | 0,177 | 0,182 | 0,186 | 0,191 | 0,196 | 0,201 | 0,206 | 0,210 |
| 30 | 0,139 | 0,143 | 0,148 | 0,153 | 0,158 | 0,163 | 0,168 | 0,173 | 0,177 | 0,182 | 0,187 | 0,192 | 0,196 | 0,201 | 0,206 |
| 35 | 0,136 | 0,140 | 0,145 | 0,150 | 0,154 | 0,159 | 0,164 | 0,169 | 0,173 | 0,178 | 0,182 | 0,187 | 0,192 | 0,196 | 0,201 |
| 40 | 0,133 | 0,137 | 0,142 | 0,147 | 0,151 | 0,156 | 0,160 | 0,165 | 0,170 | 0,174 | 0,178 | 0,183 | 0,188 | 0,192 | 0,196 |
| 50 | 0,127 | 0,132 | 0,136 | 0,141 | 0,145 | 0,149 | 0,154 | 0,158 | 0,163 | 0,167 | 0,171 | 0,175 | 0,180 | 0,184 | 0,188 |
Расчеты объемных отношений газов. тепловой эффект.
Задание №2
При взаимодействии оксида серы (IV) с 5,6 л (н.у.) кислорода в соответствии с уравнением
2SO2(г) O2(г) = 2SO3(г) Q
выделилось 19 кДж теплоты. Определите тепловой эффект реакции. (Запишите число с точностью до целых.)
Решение
Ответ: 76
Пояснение:
Рассчитаем количество вещества кислорода:
n(O2) = 5,6/22,4 = 0,25 моль
Т.е. при взаимодействии 0,25 моль кислорода с оксидом серы (IV) выделяется 19 кДж теплоты. Из представленного в условии термохимического уравнения следует, что при взаимодействии 1 моль кислорода с оксидом серы выделяется количество теплоты равное Q (тепловой эффект реакции). Тогда справедлива пропорция:
0,25 моль кислорода — 19 кДж
1 моль кислорода — Q
Исходя из этой пропорции:
0,25 ∙ Q = 1 ∙ 19
Q = 76 кДж
Таким образом, тепловой эффект реакции составляет 76 кДж.
Задание №5
Вычислите количество теплоты, которое потребуется для получения 25 г железа согласно уравнению реакции
Fe2O3(тв.) 3CO(г) = 2Fe(тв.) 3CO2(г) — 27 кДж
(Запишите число с точностью до целых.)
Решение
Ответ: 6
Пояснение:
Рассчитаем количество вещества железа:
n(Fe) = m(Fe)/M(Fe) = 25/56 = 0,4464 моль,
Согласно термохимическому уравнению при образовании 2 моль металлического железа поглощается 27 кДж теплоты. Пусть количество поглощаемой в результате образования 0,4464 моль железа равно х кДж. Тогда можно записать пропорцию:
2 моль Fe — 27 кДж
0,4464 моль Fe — x кДж
Следовательно, справедливо уравнение:
2 ∙ x = 0,4464 ∙ 27
x = 6
Таким образом, количество теплоты, которое необходимо для получения 25 г железа равно 6 кДж.
Задание №6
Вычислите объем сгоревшего согласно уравнению реакции
2C2H2(г) 5O2(г) = 4CO2(г) 2H2O(ж) 2700 кДж
ацетилена, если при этом выделилось 67,5 кДж теплоты. (Запишите число с точностью до сотых.)
Решение
Ответ: 1,12
Пояснение:
Согласно термохимическому уравнению при сгорании 2 моль ацетилена выделяется 2700 кДж теплоты. В нашем же случае выделилось 67,5 кДж. Пусть, количество вещества ацетилена, которое сгорело при этом равно х моль. Таким образом можно записать пропорцию:
2 моль C2H2 — 2700 кДж
x моль — 67,5 кДж
2 ∙ 67,5 = 2700 ∙ x
x = 0,05
Таким образом, количество вещества ацетилена равно 0,05 моль. Рассчитаем его объем:
V(C2H2) = 0,05 моль ∙ 22,4 л/моль = 1,12 л
Задание №7
В результате реакции, термохимическое уравнение которой
C6H12O6(тв.) 6O2(г) = 6CO2(г) 6H2O(г) 2800 кДж
выделилось 280 кДж теплоты. Вычислите массу глюкозы, которая окислилась в результате этой реакции. (Запишите число с точностью до целых.)
Решение
Ответ: 18
Пояснение:
Согласно термохимическому уравнению при окислении 1 моль глюкозы выделяется 2800 кДж теплоты. В нашем же случае выделилось 280 кДж теплоты. Пусть количество глюкозы, которое окислилось при этом равно x моль. Тогда можно записать следующую пропорцию:
1 моль глюкозы – 2800 кДж теплоты
x моль глюкозы – 280 кДж теплоты
Следовательно, справедливо уравнение:
1 ∙ 280 = 2800 ∙ x
Решая которое находим, что:
x = 0,1 моль, т.е. количество глюкозы составляет 0,1 моль. Рассчитаем ее массу:
m(C6H12O6) = M(C6H12O6) ∙ n(C6H12O6) = 0,1 моль ∙ 180 г/моль = 18 г.
Задание №11
В ходе реакции, термохимическое уравнение которой
CaCO3(тв.) = CaO(тв.) CO2(г) -180 кДж,
было затрачено 450 кДж теплоты. Определите объем (н.у.) получившегося при этом газа. (Запишите число с точностью до целых.)
Решение
Ответ: 56
Пояснение:
Выделившийся газ – CO2.
В соответствии с термохимическим уравнением при образовании одного моля CO2 поглощается 180 кДж теплоты. В нашем же случае поглощено 450 кДж теплоты. Обозначив количество вещества углекислого газа, образовавшегося при этом как x моль, можно записать следующую пропорцию:
1 моль CO2 – 180 кДж
x моль CO2 – 450 кДж
Следовательно, справедливо уравнение:
1 ∙ 450 = 180 ∙ x
x = 2,5, т.е. количество вещества углекислого газа составляет 2,5 моль. Рассчитаем его объем:
V(CO2) = Vm ∙ n(CO2) = 22,4 ∙ 2,5 = 56 л
Задание №13
Определите количество теплоты, которое выделится при взаимодействии 280 г негашеной извести с водой в соответствии с термохимическим уравнением реакции
CaO(тв.) H2O(ж) = Ca(OH)2(тв.) 64,8 кДж.
(Запишите число с точностью до целых.)
Решение
Ответ: 324
Пояснение:
Негашеная известь — тривиальное название оксида кальция CaO. Рассчитаем его количество вещества, зная массу:
n(CaO) = 280 г/ 56 г/моль = 5 моль,
В соответствии с термохимическим уравнением реакции при взаимодействии с водой 1 моль CaO выделяется количество теплоты равное 64,8 кДж. В нашем же случае в реакции принимает участие 5 моль оксида кальция, в связи с чем мы можем записать следующую пропорцию:
1 моль CaO — 64,8 кДж
5 моль CaO — x кДж
Следовательно, справедливо уравнение:
1 ∙ x = 5 ∙ 64,8
Решая которое находим, что:
x = 324 кДж
Таким образом, количество теплоты, которое выделится при взаимодействии 280 г негашеной извести водой составляет 324 кДж.
Задание №15
При образовании 560 мл (н.у.) газообразного аммиака в соответствии с уравнением
N2(г) 3H2(г) = 2NH3(г) Q
выделилось 1,15 кДж теплоты. Чему равен тепловой эффект этой реакции? (Запишите число с точностью до целых.)
Решение
Ответ: 92
Пояснение:
Посчитаем какое количество вещества содержится в 560 мл газообразного аммиака. Прежде всего переведем его объем из мл в л:
560 мл = 0,56 л.
Теперь, зная, что 1 моль любого газа при н.у. занимает объем равный 22,4 л, можно рассчитать количество вещества аммиака:
n(NH3) = V(NH3)/Vm = 0,56 л / 22,4 л/моль = 0,025 моль,
Из условия задания следует, что при образовании 0,025 моль (0,56 л) аммиака выделилось 1,15 кДж теплоты. В термохимическом же уравнении реакции фигурирует количество вещества аммиака равное 2 моль (коэффициент 2 перед NH3) и при этом выделяется количество тепла, равное Q (тепловой эффект реакции). Таким образом, мы можем записать следующую пропорцию:
0,025 моль — 1,15 кДж
2 моль — Q
Следовательно справедливо уравнение:
0,025 ∙ Q = 2 ∙ 1,15 кДж
Решая которое находим, что:
Q = 92 кДж
Т.е. тепловой эффект реакции равен 92 кДж.
Задание №18
В соответствии с термохимическим уравнением реакции
C(тв.) O2(газ) = CO2(газ) 393 кДж
получено 422,2 кДж теплоты. Какой объем (н.у.) кислорода при этом израсходован? (Запишите число с точностью до целых.)
Решение
Ответ: 24
Пояснение:
В соответствии с термохимическим уравнением при израсходовании на горение угля 1 моля кислорода выделяется 393 кДж теплоты. В нашем же случае выделяется 422,2 кДж. Пусть при выделении такого количества теплоты расходуется x моль кислорода. Тогда, мы можем записать пропорцию:
1 моль O2 – 393 кДж
x моль O2 – 422,2 кДж
Следовательно, справедливо уравнение:
1 ∙ 422,2 = 393 ∙ x
Решая которое находим, что:
x = 1,0743
т.е. количество вещества израсходованного кислорода составляет 1,0743 моль. Рассчитаем его объем:
V(O2) = Vm ∙ n(O2) = 22,4 ∙ 1,0743 ≈ 24 л
Задание №20
При сжигании ацетилена в соответствии с термохимическим уравнением реакции
2C2H2(г) 5O2(г) = 4CO2(г) 2H2O(ж) 930 кДж
выделилось 186 кДж теплоты. Какой объем (н.у.) кислорода затрачен на сжигание ацетилена? (Запишите число с точностью до десятых.)
Решение
Ответ: 22,4
Пояснение:
Из термохимического уравнения реакции следует, что когда в реакции принимает участие 5 моль O2 выделяется 930 кДж теплоты.
В нашем же случае выделяется 186 кДж теплоты. Пусть количество вещества кислорода, израсходованного при этом, составляет x моль. Тогда мы можем записать следующую пропорцию:
5 моль O2 – 930 кДж
x моль O2 – 186 кДж
Следовательно, справедливо уравнение:
5 ∙ 186 = 930 ∙ x
Решая которое находим, что:
x = 1
Таким образом, количество вещества кислорода составляет 1 моль. Найдем его объем:
V(O2) = n(O2) ∙ Vm = 1 ∙ 22,4 = 22,4 л