
, который можно получитпри каталитическом окислении в лаборатории 5,6л аммиака, если объемная д")
Окисление аммиака до оксида азота (ii) — киберпедия
При окислении аммиака кислородом воздуха на катализаторе возможно протекание следующих реакций:
1) 4NH3 5O2 = 4NO 6H2O – ΔH; ΔH = 907,3 кДж;
2) 4NH3 4O2 = 2N2O 6H2O – ΔH; ΔH = 1104,9 кДж;
3) 4NH3 3O2 = 2N2 6H2O – ΔH; ΔH = 1269,1 кДж;
а также реакция с участием образующегося оксида азота (II):
4NH3 6NO = 5N2 6H2O – ΔH; ΔH = 110 кДж.
Все реакции практически необратимы, поэтому направление процесса окисления определяется соотношением скоростей этих реакций. Из трех основных реакций окисления аммиака реакция 3 термодинамически наиболее вероятна, так как протекает с максимальным выделением тепла. Поэтому, в отсутствии катализатора окисление аммиака идет преимущественно до элементарного азота. Для ускорения целевой реакции окисления до оксида азота (II) применяют селективно действующие катализаторы. В современных установках используют платиновые катализаторы в виде пакета сеток из сплава платины с 7,5 % родия, или двухступенчатые катализаторы в виде слоя таблетированной смеси оксидов железа (III) и хрома (III). Введение родия повышает механическую прочность и уменьшает потери платины за счет ее уноса тоном газа. Поверхность подобных катализаторов достигает 1,5 м2/м3 объема.
Механизм гетерогенного каталитического окисления аммиака состоит из следующих последовательных стадий:
· диффузия молекул аммиака и кислорода из газовой фазы к поверхности катализатора;
· активированная адсорбция молекул кислорода на поверхности катализатора с образованием промежуточного соединения;
· хемосорбция молекул аммиака и образование комплекса;
· разложение комплекса с регенерацией катализатора и образованием молекул оксида азота (II) и воды;
· диффузия продуктов реакции с поверхности катализатора в газовую фазу.
Схема действия катализатор представлена на рис.:
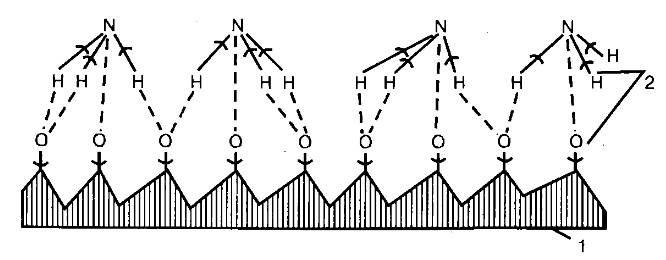
Рис. Схема окисления аммиака на катализаторе:
сплошные линии – ранее возникшие связи;
пунктирные – вновь образующиеся связи.
1 – катализатор, 2 – места разрыва связей.
Определяющей стадией всего процесса окисления является скорость диффузии кислорода к поверхности катализатора. Следовательно, каталитическое окисление аммиака на платиновом катализаторе протекает преимущественно в диффузионной области, в отличие от окисления на окисном катализаторе, которое идет в кинетической области.
Платиновые катализаторы весьма чувствительны к каталитическим ядам, содержащимся в аммиаку и воздухе, образующим аммиачно-воздушную смесь (АмВС). Фосфористый водород вызывает его необратимое, а ацетилен, сероводород и органические соединения серы обратимое отравление. Так как вследствие этого активность катализатора снижается, его периодически регенерируют промывкой соляной или азотной кислотой.
В процессе работы поверхность катализатора разрушается и частицы его уносятся с потоком газа. Эрозия катализатора тем больше, чем выше температура, давление и объемная скорость газа, проходящего через катализатор. Для систем, работающих под высоким давлением, унос катализатора составляет 0,3 – 0,4 г на тонну азотной кислоты.
В присутствии платиновых катализаторов селективность процесса окисления аммиака до оксида азота (II):
sNO = m NO / (m NO m N O m N )
составляет 0,95 – 0,98 дол. ед. В этих условиях скорость окисления до оксида азота (II) описывается уравнением:
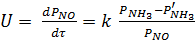 ,
,
где: 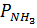 — парциальное давление аммиака, окисляемого до оксида азота (II),
— парциальное давление аммиака, окисляемого до оксида азота (II),
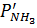 — парциальное давление аммиака, окисляемого до оксида азота (I) и элементарного азота,
— парциальное давление аммиака, окисляемого до оксида азота (I) и элементарного азота,
k – константа скорости.
Энергия активации этой реакции составляет 33,494 кДж/моль.
Из двух, конкурирующих с целевой реакцией окисления аммиака, наиболее опасной является реакция, приводящая к образованию элементарного азота. Скорость обеих реакций может быть описана общим для гетерогенных реакций уравнением:
U = k M ∙ F KT ∙ ΔC
и зависит от таких параметров процесса как температура (через k M), давление и состав АмВС, то есть отношение кислород : аммиак (через ΔС), время контактирования, то есть время пребывания АмВС в зоне катализатора. Влияние этих факторов на скорость окисления аммиака до оксида азота (II) и до азота и, следовательно, выход продуктов окисления, различно.
Влияние температуры
Повышение температуры способствует увеличению скорости реакций и коэффициента диффузии аммиака в смеси и, поэтому, является наиболее эффективным средством, увеличения скорости процесса, протекающего преимущественно в диффузионной области. Это подтверждается термодинамическими данными.
| Реакция | ΔН, кДж/моль | ΔG, кДж/моль | |
| 298 ºК | 1173 ºК | ||
| 4NH3 5O2=4NO 6H2O | -226,0 | -246,2 | -414,6 |
| 4NH3 3O2=2N2 6H2O | -317,2 | -326,9 | -335,2 |
Из таблицы следует, что вероятность реакции окисления до оксида азота (II) с повышением температуры возрастает почти вдвое, а реакции окисления до азота почти не изменяется.
Влияние состава АмВС
Соотношение аммиака и кислорода в газовой смеси влияет на температурный режим и общую скорость процесса в том случае, если лимитирующей в нем является химическая реакция, то есть процесс протекает в кинетической области. При стехиометрическом соотношении компонентов в АмВС степень превращения аммиака в оксид азота (II) не превышает 0,65 дол.ед. Для увеличения выхода оксида азота (II) процесс ведут при отношении О2 : NH3 = 1,8 – 2,0, что соответствует содержанию в АмВС 0,095 – 0,105 об. долей аммиака и 0,18 – 0,19 об. долей кислорода. Избыток кислорода используется на стадии доокисления оксида азота (II), а указанный состав АмВС обеспечивает автотермичность процесса окисления и лежит за пределом взрывчатости АмВС.
Влияние давления
Повышение давления ускоряет процесс окисления аммиака за счет увеличения концентрации реагентов и производительности катализатора, что позволяет сократить размеры аппаратуры. При этом, однако, снижается выход оксида азота (II) и увеличивается эрозия и унос катализатора, что удорожает продукцию. Так, если при атмосферном давлении (105 Па) унос катализатора не превышает 0,05 г на тонну азотной кислоты, то при давлении 0,8 МПа он достигает 0,4 г/тонну.
§
Скорость каталитического окисления аммиака до оксида азота (II) весьма высока. За десятитысячные доли секунды степень превращения составляет 0,97 – 0,98 дол.ед. при атмосферном давлении и 0,98 – 0,96 при давлении 0,8 – 1,0 МПа. Время контактирования может быть рассчитано из формулы:
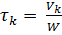 ,
,
где: Vk – объем катализатора, определяемый в случае платинового катализатора числом сеток в пакете,
W – объемная скорость АмВС.
Время контактирования зависит от природы катализатора и составляет: для платиновых катализаторов около 10-2 с. Увеличение времени контактирования, то есть снижение объемной скорости АмВС приводит к развитию реакции окисления аммиака до элементарного азота.
Оптимальный режим процесса на этой стадии должен обеспечить селективность окисления аммиака, минимальные потери катализатора вследствие его уноса и автотермичность процесса. Этим требованиям удовлетворяют следующие условия: температура 800 ºС, давление 0,1 – 1,0 МПа, молярное отношение О2 : NH3 = 1,8 – 2,0, время контактирования 1-2∙10-4 сек.
Для соблюдения этих условий исходная АмВС должна иметь состав: аммиак 0,10 – 0,115 об.дол., кислород 0,18 – 0,19 об.дол., азот 0,70 – 0,72 об.дол.
При использовании АмВС такого состава нитрозные газы, выходящие из контактного аппарата, содержат от 0,08 до 0,11 об.дол. оксида азота (II).
Катализаторы
Превосходство платины по активности и селективности над всеми другими видами катализаторов было показано в 1902 г. Оствальдом. Характерно, что активность к реакции окисления аммиака проявляет подавляющее большинство металлов и их соединений, но высокий выход N0 (выше 90%) обеспечивают очень немногие из них. Обладая высокой активностью и селективностью, платина имеет низкую температуру зажигания 195 ºС, хорошую пластичность, тягучесть. Недостаток платины — ее быстрое разрушение при высоких температурах под воздействием больших скоростных потоков реагентов и катализаторных ядов. Это приводит к потерям дорогостоящего катализатора и снижению выхода N0, что и явилось причиной поисков каталитически активных сплавов платины с другими металлами. Проведенные промышленные испытания показали стабильную работу катализаторов из платины с добавками палладия, а также из тройного сплава Pt-Rh-Pd. Наибольшее распространение получили следующие катализаторы окисления аммиака: Pt 4% Pd 3,5%Rh — для работы при атмосферном давлении и Pt 7,5% Rh — при повышенном давлении.
Используемые для контактного окисления NH3 катализаторы изготавливают в виде сеток. Такая форма катализатора удобна в эксплуатации, связана с минимальными затратами металла, позволяет применять наиболее простой и удобный в эксплуатации тип контактного аппарата. В нашей стране применяются сетки из проволоки диаметром 0,09 мм, размер стороны ячейки 0,22 мм, число ячеек на 1 см длины — 32, на 1см2 – 1024.
Платинородиевые (ГИАП-1) и платинородиевопалладиевые (сплав № 5) катализаторы весьма чувствительны к ряду примесей, которые содержатся в аммиаке и воздухе. К таким примесям относятся гидриды фосфора и мышьяка, фтор и его соединения, дихлорэтан, минеральные масла, ацетилен, диоксид серы, сероводород и др. Наиболее сильными ядами катализатора являются соединения серы и фтора. Примеси заметно снижают селективность катализатора, способствуют увеличению потерь платины. Для поддержания стабильной степени конверсии аммиака необходима тщательная очистка аммиачно-воздушной смеси и от механических примесей, особенно от оксидов железа и пыли железного катализатора синтеза аммиака. Пыль и оксиды железа, попадая на катализаторные сетки, засоряют их, уменьшая поверхность соприкосновения газов с поверхностью катализатора, и снижают степень окисления аммиака.
Чистоту исходных веществ в производстве азотной кислоты обеспечивают двумя путями – дальним забором воздуха и усовершенствованием систем очистки воздуха и аммиака.
В процессе реакции окисления аммиака поверхность платиноидных сеток сильно разрыхляется. эластичные нити сеток становятся хрупкими, поверхность сетки увеличивается примерно в 30 раз. Сначала это ведет к повышению каталитической активности катализатора, а затем к разрушению сеток. Практикой установлены следующие сроки работы катализаторных сеток: для работы под атмосферном давлении – до 14 мес., под давлением 0,73 МПа – 8-9 мес.
§
Нитрозные газы, полученные на стадии окисления аммиака, содержат оксид азота (II), азот, кислород и пары воды. При окислении оксида азота (II) в оксид азота (IV) в этой системе протекают три параллельных реакции:
1) 2NO O2 2NO2 – ΔH, ΔH = 112,3 кДж,
2) 2NO2 N2O4 – ΔH, ΔH = 57,0 кДж,
3) NO2 NO N2O3 – ΔH, ΔH = 40,0 кДж.
Все эти реакции обратимы, протекают в гомогенной системе с выделением тепла и уменьшением объема. Вследствие этого понижение температуры и повышение давления сдвигает равновесие их вправо.
Константа равновесия первой реакции окисления оксида азота (II) выражается уравнением:
 ,
,
и сильно зависит от температуры.
| tºC | ||||||
| Kp | 1,24∙1014 | 1,82∙108 | 8,5∙104 | 2,1∙10-3 | 5,6∙10-3 | 1,5∙10-3 |
Из таблицы следует, что при температурах ниже 100 ºС равновесие первой реакции почти полностью сдвинуто в сторону образования оксида азота (IV). При повышении температуры оно сдвигается влево и выше 700 ºС образования оксида азота (IV) практически не происходит. Так как нитрозные газы выходят из реактора при температуре около 800 ºС, в них оксид азота (IV) газы необходимо охладить ниже 100 ºС.
Обычно переработку нитрозных газов ведут при 10 – 50 ºС. В этих условиях часть оксида азота (IV) димеризуется в тетроксид N2O4. Степень димеризации его существенно зависит от температуры. При температуре выше 150 ºС равновесие второй реакции почти полностью сдвинуто влево и тетроксид азота в газе практически отсутствует. Даже при -20 ºС степень димеризации оксида азота (IV) не превышает 92%.
Скорости первой и второй реакций различны, поэтому соответствующие равновесия устанавливаются не одновременно. Первая реакция окисления протекает с меньшей скоростью, поэтому скорость всего процесса на этой стадии производства определяется именно скоростью окисления оксида азота (II), которая выражается общим для гомогенных реакций уравнением:
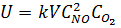 .
.
Для этой реакции характерна аномальная зависимость ее скорости от температуры. Она ускоряется при понижении температуры и почти полностью прекращается с повышением температуры до определенного предела. Это объясняется особым механизмом окисления оксида азота (II) в оксид азота (IV), которое протекает в две стадии через образование промежуточного продукта – димера оксида азота (II):
2 NO N2O2 – ΔH,
N2O2 O2 2NO2 – ΔH.
Реакция образования димера обратима, протекает с выделением тепла и значительно быстрее, чем реакция его последующего окисления. Поэтому при повышении температуры равновесие реакции образования димера сдвигается влево и равновесная концентрация димера в газе понижается. Так как скорость реакции окисления димера:
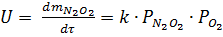
зависит от его концентрации, то уменьшение скорости при повышении температуры вызывает снижение скорости окисления димера и, следовательно, оксида азота (II) до оксида азота (IV).
Скорость реакции димеризации оксида азота (IV) в тетроксид весьма высока, поэтому равновесие второй реакции устанавливается практически мгновенно и соотношение оксидов NO2 : N2O4 определяется условиями этого равновесия, установившегося в газе.
Таким образом, понижение температуры и повышение давления в нитрозном газе способствует окислению оксида азота (II) в оксид азота (IV) и димеризации последнего.
Абсорбция оксида азота (IV)
Нитрозные газы, поступающие на абсорбцию, представляют сложную смесь различных оксидов азота (NO2, N2O4, NO, N2O), элементарного азота, кислорода и паров воды. Их состав зависит от условий окисления, то есть от состояния системы, описываемого тремя реакциями.
Все оксиды азота, входящие в состав нитрозных газов, нерастворимы в воде, но, за исключением оксида азота (II), взаимодействуют с ней. Поглощение их водой сопровождается химической реакцией хемосорбции, протекающей в системе «газ-жидкость», описываемой уравнениями:
2NO2 H2O HNO3 HNO2 – ΔH, ΔH = 116 кДж,
N2O4 H2O HNO3 HNO2 – ΔH, ΔH = 59 кДж
и распада нестойкой азотистой кислоты по уравнению:
3HNO2 HNO3 2NO H2O ΔH, ΔH = 76 кДж.
Суммируя эти уравнения, получаем итоговые уравнения поглощения оксидов азота водой:
3NO2 г H2O ж 2HNO3 ж NO г – ΔH, ΔH = 136 кДж,
3N2O4 г H2O ж 4HNO3 ж 2NO г – ΔH, ΔH = 101 кДж.
Из этих уравнений следует, что при абсорбции из трех моль оксида азота (IV) образуется два моля азотной кислоты и один моль оксида азота (II), который возвращается в цикл и снова окисляется до оксида азота (IV).
Механизм образования азотной кислоты при адсорбции оксида азота (IV) водой, а затем образующейся водной азотной кислотой, заключается в том, что оксид азота (IV) диффундирует через пограничный слой газа к поверхности жидкости и абсорбируется ею. При этом оксид азота (IV) реагирует с водой в первой реакции со скоростью, превышающей скорость диффузии и скорость реакции разложения азотистой кислоты в третьей реакции. Образующийся оксид азота (II) выделяется в газовую фазу, где окисляется кислородом до оксида азота (IV).
Скорость процесса абсорбции оксида азота (IV) водой описывается уравнения для гетерогенных процессов:
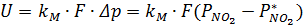 ,
,
где Δp – движущая сила абсорбции,
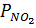 – парциальное давление NO2 в газовой фазе,
– парциальное давление NO2 в газовой фазе,
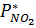 – равновесное давление NO2 у поверхности водного раствора азотной кислоты.
– равновесное давление NO2 у поверхности водного раствора азотной кислоты.
С повышением концентрации кислоты в процессе абсорбции возрастает равновесное давление оксида азота (IV) и снижается движущая сила процесса. Вследствие этого процесс абсорбции замедляется.
Состояние системы «NO2 – HNO3 – H2O» и, следовательно, концентрация получаемой азотной кислоты зависит от температуры, давления, парциального давления оксида азота (IV) в поглощаемой газовой смеси и концентрации образовавшейся кислоты. При понижении температуры и концентрации кислоты и повышения давления степень абсорбции оксида азота (IV) водной азотной кислотой возрастает, при том тем интенсивнее, чем выше концентрация его в нитрозных газах. При атмосферном давлении и температуре 25 ºС абсорбция оксида азота практически прекращается, когда концентрация кислоты достигнет 0,65 мас.долей.
Таким образом, возможность получения азотной кислоты концентрацией более 0,65 мас.дол. объективно ограничена температурой и давлением процесса абсорбции и содержанием оксида азота (IV) в нитрозных газах. В реальных условиях производства при температуре 40 ºС, давлении 0,1 МПа и понижении содержания оксида азота вследствие его поглощения из газа концентрация получаемой кислоты не превышает 0,5 мас.дол. Получение азотной кислоты более высокой концентрации требует иной технологии.
Степень абсорбции оксида азота (IV) непосредственно связана с абсорбционным объемом аппаратуры. Повышение степени абсорбции требует, особенно, в конце процесса, значительного увеличения абсорбционного объема. Так, если степень абсорбции, равная 0,92 дол.ед., может быть достигнута при Vаб = 22 м3/т кислоты, то для повышения ее до 0,98 дол.единицы, то есть на 6,5% абсорбционный объем должен быть увеличен до 70 м3/т. Так как увеличение абсорбционного объема вызывает резкое возрастание капитальных затрат, то экономически более выгодно не добиваться степени абсорбции выше 0,98 дол.ед., а поглощать остатки оксида азота (IV) в отходящих газах щелочными поглотителями с последующим окислением образовавшегося нитрита натрия концентрированной азотной кислотой и возвращением оксида азота (II) в цикл (инверсия оксида азота (II)):
2NO2 Na2CO3 = NaNO2 NaNO3 CO2,
3NaNO2 2HNO3 = 3NaNO3 2NO H2O.
§
Процесс синтеза азотной кислоты из аммиака протекает в несколько стадий, каждая из которых включает несколько химических реакций. Для количественных расчетов процесса, в частности, определения расходных коэффициентов и общего выхода азотной кислоты на исходное сырье, целесообразно пользоваться итоговым уравнением, полученным суммированием уравнений реакций каждой стадии после соответствующих операций деления и умножения их:
4NH3 5O2 = 4NO 6H2O x 2
2NO O2 = 2NO2 x 6
3NO2 H2O = 2HNO3 NO x 4
8NH3 16O2 = 8HNO3 8H2O : 8
NH3 2O2 = HNO3 H2O
Охрана окружающей среды
Неуклонный рост производства азотной кислоты тесно связан с увеличением о6ъемаотходяших газов, а следовательно, с ростом количества выбрасываемых в атмосферу оксидов азота. Оксиды азота очень опасны для любых живых организмов. Некоторые растения повреждаются уже через 1 ч пребывания в атмосфере, содержащей 1 мг оксидов в 1 м3 воздуха. Оксиды азота вызывают раздражение слизистой оболочки дыхательных путей, ухудшение снабжения тканей кислородом и другие нежелательные последствия. Хвостовые газы производства азотной кислоты содержат после абсорбционных колонн от 0,05 до 0,2% оксидов азота, которые по санитарным требованиям без дополнительной очистки запрещено выбрасывать в атмосферу.
Радикальное решение проблем очистки хвостовых газов – каталитическое восстановление оксидов азота снижает их содержание в очищенном газе до 0,001-0,005%. что обеспечивает санитарные нормы по содержанию оксидов азота в приземном слое воздуха при мощностях производства кислоты до 1 млн т/год, сосредоточенных в одной точке и при высоте выброса 100-150 м. На ряде существующих предприятий по производству азотной кислоты под давлением применен метод очистки, в котором восстановителем является природный газ, а в качестве катализатора используется палладий на носителе, в качестве второго слоя катализатора — носитель (оксид алюминия). На катализаторе протекают следующие реакции:
CH4 202 CO2 2Н2О;
CH4 2NO2 N2 2Н2О СО2;
CH4 4NO 2N2 2Н2О СО2.
Метод обеспечивает снижение содержания оксидов азота в выхлопных газах до 0,005%. Теплота реакции используется для получения пара. Данный метод очистки органически связан с технологией производства азотной кислоты. Он нашел применение в агрегате, работающем под давлением 0,716 МПа, и в aгpeгaтe AK-72.
В качестве восстановителя вместо природного газа применяют также аммиак. Этот метод характеризуется тем, что на алюмованадиевом катализаторе аммиак реагирует только с оксидами азота, т. е. обеспечивает селективное восстановление:
4NH3 6NO 5N2 6H2О;
8NH3 6NO2 7N2 12H2О.
Одним из наиболее реальных способов утилизации оксидов азота, обеспечивающих санитарные нормы содержания оксидов азота в приземном слое атмосферы после рассеивания их из выхлопной трубы, является адсорбционно-десорбционный метод, в котором используется непрерывно циркулирующий сорбент (силикагель). Разработаны способы адсорбции на молекулярных ситах, промывки кислым раствором мочевины и другими промывными жидкостями. На современных установках получения азотной кислоты нет постоянных источников сточных вод. Эти установки потребляют большое количество оборотной охлаждающей воды. Растворы, периодически сливаемые из насосов и другого оборудования, например при проведении ремонта, собирают в приямок и нейтрализуют.


Схема производства азотной кислоты под давлением 0,716 МПа с приводом компрессора от газовой турбины:
1 – фильтр воздуха; 2 – реактор каталитической очистки; 3 – топочное устройство; 4 – подогреватель метана; 5 – подогреватель аммиака; 6 – смеситель аммиака и воздуха с пролитовым фильтром; 7 – холодильник-конденсатор; 8 – сепаратор; 9 – абсорбционная колонна; 10 – продувочная колонна; 11 – подогреватель отходящих газов; 12 – подогреватель воздуха; 13 – сосуд для окисления нитрозных газов; 14 – контактный аппарат; 15 – котел‑утилизатор; 16,18 – двухступенчатый турбокомпрессор; 17 – газовая турбина
§
Блок-схема
Рис. Структурная блок-схема производства азотной кислоты:
1 – блок подготовки сырья; 2 – блок окисления аммиака; 3 – блок абсорбции нитрозных газов.
Условно постоянная информация для расчета
G – масса потока;
V – объём потока;
N – количество молей потока;
g – доля компонентов в потоке: нижний индекс номер потока; верхний – компонент.
Составы потоков
| № потока | Индекс потока | Ед. измер. | Содержание компонентов | |||||
| NH3 | O2 | N2 | NO | H2O | HNO3 | |||
| кмоль | 100% | |||||||
| кмоль | 21% | 79% | ||||||
| кмоль | 9,4% | 19,0% | 71,5% | |||||
| кмоль | кмоль | кмоль | кмоль | кмоль | ||||
| кмоль | кмоль | |||||||
| кмоль | кмоль | кмоль | ||||||
| кг | 44% | 59% | ||||||
| кмоль | 3,0% | кмоль | кмоль | 2,6% |
Балансовая математическая модель
Составление системы уравнений
4NH3 5O2 = 4NO 6H2O ΔH1
2NO O2 2NO2 ΔH2
2NO 1,5O2 H2O = 2HNO3 ΔH3
СН4 4NO = CO2 2N2 2H2O
Уравнения для блока контактирования:
По оксиду азота:
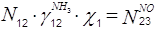
По азоту:
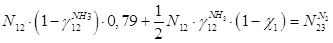
По кислороду:
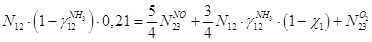
По воде:
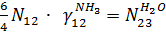
Уравнения для блока абсорбции:
По оксиду азота:
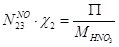
По воде:
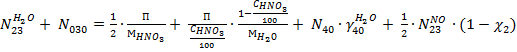
По кислороду:
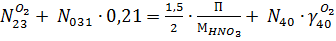
Уравнение для потока выхлопных газов:
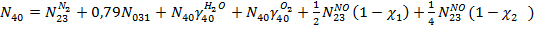
По метану:

Подготовка системы для решения на ЭВМ
Соответствие переменных потокам
Матрица коэффициентов
| № ур-я | ai при xi | Свободный член bi | ||||||||
| х1 | х2 | х3 | х4 | х5 | х6 | х7 | х8 | х9 | ||
| 0,09 | –1 | |||||||||
| 0,717 | –1 | |||||||||
| 0,186 | –1,25 | –1 | ||||||||
| –0,14 | ||||||||||
| 47,62 | ||||||||||
| -0,01 | –0,025 | 154,76 | ||||||||
| –0,030 | 0,21 | 35,71 | ||||||||
| –0,03 | –1 | 0,945 | -0,79 | |||||||
| -0,005 |
| Обратная матрица | |||||||||
| 11,11 | 0,00 | 0,00 | 0,00 | 11,11 | 0,00 | 0,00 | 0,00 | 0,00 | |
| 0,00 | 0,00 | 0,00 | 0,00 | 1,00 | 0,00 | 0,00 | 0,00 | 0,00 | |
| 7,97 | -1,00 | 0,00 | 0,00 | 7,97 | 0,00 | 0,00 | 0,00 | 0,00 | |
| 2,07 | 0,00 | -1,00 | 0,00 | 0,82 | 0,00 | 0,00 | 0,00 | 0,00 | |
| 1,56 | 0,00 | 0,00 | 1,00 | 1,56 | 0,00 | 0,00 | 0,00 | 0,00 | |
| -1,55 | -0,03 | 0,11 | -1,00 | -1,40 | 1,00 | 0,11 | 0,03 | 0,00 | |
| 0,23 | -1,20 | 4,52 | 0,00 | 5,91 | 0,00 | 4,52 | 1,20 | 0,00 | |
| -9,81 | -0,17 | 5,41 | 0,00 | -3,05 | 0,00 | 5,41 | 0,17 | 0,00 | |
| 0,00 | 0,00 | 0,00 | 0,00 | 0,03 | 0,00 | 0,00 | 0,00 | 1,00 |
| х1 | ||
| х2 | ||
| х3 | ||
| х4 | ||
| х5 | ||
| х6 | ||
| х7 | 443,2 | |
| х8 | ||
| x9 |
| х1 | |
| х2 | |
| х3 | |
| х4 | |
| х5 | |
| х6 | |
| х7 | 443,2 |
| х8 | |
| x9 |
Материальный баланс ХТС
Расчет
1.Введено
N(NH3) =N12 ∙ 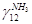 = 529,1 ∙ 0,094 =49,74 кмоль
= 529,1 ∙ 0,094 =49,74 кмоль
m (NH3) = N(NH3) ∙ M(NH3) = 49,74 ∙ 17 = 845,58 кг
N(N2) = N12 ∙ (1 — ) ∙ 0,79 = 529,1 ∙ (1 – 0,094) ∙ 0,79 = 378,7 кмоль
m (N2) = N(N2) ∙ M(N2) = 378,7 ∙ 28 = 10603,6 кг
N(O2) = N12 ∙ (1 — ) ∙ 0,21 = 529,1 ∙ (1 – 0,094) ∙ 0,21 = 100,7 кмоль
m (O2) = N(O2) ∙ M(O2) = 100,7 ∙ 32 = 3222,4 кг
m (возд.) = m (O2) m (N2) = 3222,4 10603,6 = 13826 кг
2. Получено
m (HNO3)56 = 1500 кг (по условию)
m (HNO3) = m (HNO3)56 ∙ СHNO3 = 1500 ∙ 0,56 = 840 кг
m (H2O) = m (HNO3)56 — m (HNO3) = 1500 – 840 = 660 кг
N(H2O)ВГ = N40 ∙ 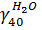 = 443,2 ∙ 0,026 = 11,52 кмоль
= 443,2 ∙ 0,026 = 11,52 кмоль
m (H2O)ВГ = N(H2O)возд. ∙ М(H2O) = 11,52 ∙ 18 = 207,36 кг
N(NO)ВГ = NNO23 ∙ (1 — χ2) = 47,6 ∙ (1 – 0,95) = 2,38 кмоль
m (NO)BГ = N(NO)ВГ ∙ М(NO) = 2,38 ∙ 30 = 71,4 кг
N(O2)ВГ = N40 ∙ 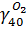 = 443,2 ∙ 0,03 = 13,3 кмоль
= 443,2 ∙ 0,03 = 13,3 кмоль
m (O2)ВГ = N(O2)ВГ ∙ M(O2) = 13,3 ∙ 32 = 426 кг
N(N2)ВГ = N12 ∙ (1 — ) ∙ 0,79  N12 ∙ ∙ (1 — χ2) =
N12 ∙ ∙ (1 — χ2) =
=529,1 ∙ (1 – 0,094) ∙ 0,79 529,1 ∙ 0,094 ∙ (1 – 0,95) = 379,9 кмоль
m (N2)ВГ = N(N2)ВГ ∙ M (N2) = 379,9 ∙ 28 = 10637,2 кг
Материальный баланс химико-технологической системы производства азотной кислоты на 1500 кг
| Введено | Получено | ||||
| Статья прихода | Масса, кг | % | Статья расхода | Масса, кг | % |
| Аммиак | 845,58 | 5,7 | 56% -я азотная к-та: | ||
| Воздух: | Азотная кислота | 11,8 | |||
| Кислород | 3222,4 | 21,7 | Вода | 9,3 | |
| Азот | 10603,6 | 71,3 | Выхлопные газы: | 11271,86 | |
| Вода | 207,36 | 1,4 | Оксид азота | 28,5 | 0,2 |
| Азот | 74,4 | ||||
| Кислород | 3,0 | ||||
| Вода | 207,36 | 1,5 | |||
| Всего: | 14878,94 | Всего: | 14271,86 |
Невязка: 14878,94 – 14271,86 = 607,08кг
Расчет основных технологических показателей процесса
Расходные коэффициенты по сырью (аммиаку):
· Теоретический расходный коэффициент:
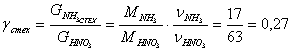
· Практический расходный коэффициент:
γ =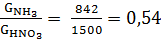
Конверсия аммиака.
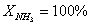 (по условию).
(по условию).
Выход.
h = 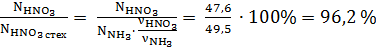
Селективность.
φ = 96,2 %,поскольку степень превращения равна 100%.
Выводы
Исключительное значение азотной кислоты для многих отраслей народного хозяйства и оборонной техники и большие объемы производства обусловили интенсивную разработку эффективных и экономически выгодных направлений совершенствования азотнокислотного производства. К таким направлениям относятся:
— создание систем высокой единичной мощности (до 400 тыс. т/год), работающих по комбинированной схеме;
— разработка высокоактивных избирательно действующих неплатиновых катализаторов окисления аммиака;
— возможно более полное использование энергии сжатых отходящих газов и низкопотенциальной теплоты процессов путем создания полностью автономных энерготехнологических схем;
— создание замкнутого оборота охлаждающей воды;
— решение проблемы очистки отходящих газов с утилизацией оксидов азота путем внедрения адсорбционно-десорбционного метода очистки на силикагеле и цеолитах;
— возможно более полное удаление остатков оксидов азота из отходящих газов с использованием в качестве восстановителей горючих газов и аммиака.
Список использованной литературы
1. Общая химическая технология: Учеб. для вузов/А.М. Кутепов, Т.И. Бондарева, М.Г. Беренгартен — 3-е изд., перераб. – М.: ИКЦ «Академкнига», 2003. – 528 с.;
2. Соколов Р.С. Химическая технология: Учеб. пособие для студ. высш. учеб. заведений: В 2 т. – М.: Гуманит. изд. центр ВЛАДОС, 2000. – Т. 1: Химическое производство в антропогенной деятельности. Основные вопросы химической технологии. Производство неорганических веществ. – 368 с.;
3. Кононова Г.Н., Сафонов В.В., Егорова Е.В. Расчет материального баланса химико-технологических систем интегральным метод ом. – М.: ИПЦ МИТХТ им. М.В. Ломоносова, 2007. — 30 с.;
Сборник заданий и методических указаний по расчету материального баланса химико-технологических производств. Учебно-методическое пособие. /Авт: Брук Л.Г., Егорова Е.В., Кононова Г.Н., Сафонов В.В., Смирнова С.Н., Чабан Н.Г., Шварц А.Л. Под ред. Брука Л.Г., Кононовой Г.Н., Сафонова В.В. Изд. 3-е, перераб. – М.: ИПЦ МИТХТ им. М.В. Ломоносова, 2008. – 72 с.